DrugWonks on Twitter
Tweets by @PeterPittsDrugWonks on Facebook
CMPI Videos

Video Montage of Third Annual Odyssey Awards Gala Featuring Governor Mitch Daniels, Montel Williams, Dr. Paul Offit and CMPI president Peter Pitts

Indiana Governor Mitch Daniels

Montel Williams, Emmy Award-Winning Talk Show Host

Paul Offit, M.D., Chief of the Division of Infectious Diseases and the Director of the Vaccine Education Center at the Children’s Hospital of Philadelphia, for Leadership in Transformational Medicine

CMPI president Peter J. Pitts

CMPI Web Video: "Science or Celebrity"
Tabloid Medicine

Check Out CMPI's Book
A Transatlantic Malaise
Edited By: Peter J. Pitts
Download the E-Book Version Here
CMPI Events

Donate

CMPI Reports

Blog Roll
AHRP
Better Health
BigGovHealth
Biotech Blog
BrandweekNRX
CA Medicine man
Cafe Pharma
Campaign for Modern Medicines
Carlat Psychiatry Blog
Clinical Psychology and Psychiatry: A Closer Look
Conservative's Forum
Club For Growth
CNEhealth.org
Diabetes Mine
Disruptive Women
Doctors For Patient Care
Dr. Gov
Drug Channels
DTC Perspectives
eDrugSearch
Envisioning 2.0
EyeOnFDA
FDA Law Blog
Fierce Pharma
fightingdiseases.org
Fresh Air Fund
Furious Seasons
Gooznews
Gel Health News
Hands Off My Health
Health Business Blog
Health Care BS
Health Care for All
Healthy Skepticism
Hooked: Ethics, Medicine, and Pharma
Hugh Hewitt
IgniteBlog
In the Pipeline
In Vivo
Instapundit
Internet Drug News
Jaz'd Healthcare
Jaz'd Pharmaceutical Industry
Jim Edwards' NRx
Kaus Files
KevinMD
Laffer Health Care Report
Little Green Footballs
Med Buzz
Media Research Center
Medrants
More than Medicine
National Review
Neuroethics & Law
Newsbusters
Nurses For Reform
Nurses For Reform Blog
Opinion Journal
Orange Book
PAL
Peter Rost
Pharm Aid
Pharma Blog Review
Pharma Blogsphere
Pharma Marketing Blog
Pharmablogger
Pharmacology Corner
Pharmagossip
Pharmamotion
Pharmalot
Pharmaceutical Business Review
Piper Report
Polipundit
Powerline
Prescription for a Cure
Public Plan Facts
Quackwatch
Real Clear Politics
Remedyhealthcare
Shark Report
Shearlings Got Plowed
StateHouseCall.org
Taking Back America
Terra Sigillata
The Cycle
The Catalyst
The Lonely Conservative
TortsProf
Town Hall
Washington Monthly
World of DTC Marketing
WSJ Health Blog
DrugWonks Blog
India’s Efforts to Aid Poor Worry Drug Makers (New York Times, December 30, 2013) points to the price of cancer medications as the sole impediment to access. Not so.
Price is one variable, but it is not the only one.
Less than 10% of India's 45+ million citizens with cardiovascular disease get even the most common medicines like diuretics and statins. Yet there are 10,500 licensed Indian drug manufacturers producing hundreds of generic anti-hypertensives and other CVD products on the market in India.
While the Times’ story shares a sad tale, the plural of “anecdote” isn’t “data.” The truth is much more complicated and the issue of local production vs. patent protection is a red herring. Almost every drug on the WHO’s list of essential drugs is off-patent – and yet patients in India (and almost every nation in the developing world) lacks access due to poorly run government programs, failing domestic infrastructure, dearth of healthcare providers and, worst of all, lack of diagnosis.
Also, once a lifesaving drug or treatment exists, it’s seductively easy to take it for granted. We sometimes forget the years of toil these things take to develop; the millions spent to bring a new drug or treatment from theory to actuality. As Abraham Lincoln wrote, patents “add the fuel of interest to the passion of genius.” Read More & Comment...The FTC and biosimilars
Instead of playing name game, it should choose clarity
By Peter Pitts
The Federal Trade Commission (FTC) has started to meddle on a health issue that probably isn’t on the radar of more than a couple of health-policy wonks, but it should be of interest to anyone who treats patients or cares about patient safety.
It’s about a class of medicines called “biosimilars,” copycat versions of innovator biologics — or medicines made from living cells. Most people aren’t even familiar with biologics, let alone biosimilars. Biologics are the fastest-growing segment of the pharmaceutical industry and are primarily used to treat life-threatening or difficult-to-target diseases like cancer, diabetes, multiple sclerosis, lupus, Crohn’s disease, rheumatoid arthritis and epilepsy. Many people aren’t familiar with them yet and only know about traditional chemical compounds, which are vastly different.
Why is the FTC involved? Good question. The FTC’s interest here would be to ensure that “anti-competitive” or “deceptive practices” don’t compromise consumers’ ability to know which product he or she is prescribed, and to ensure it’s the one he or she actually receives. That’s because that’s what “informed consumer choice” is all about — right?
Not in this case. The issue the FTC is actually focusing on is whether any and all biosimilars that follow a particular “reference product” (the innovator biologic) ought to have the exact same name as that reference product. And all current indications point to the FTC is about to concur that they should.
This is no trivial issue. It is a fact that no two biologic products produced by different manufacturers will be the same. A biosimilar can only resemble its reference product. Therefore, how biologics are named will directly impact clarity of information around which products a patient has been using. Greater clarity will obviously occur if biologics and biosimilars have distinguishable names, and that clarity will enable better safety monitoring, “adverse event” reporting and timeliness in managing adverse events if they occur, and can even help us better understand which products work better for certain patients and specific subpopulations.
Let’s employ some common sense here. Suppose parents were to give birth to a set of fraternal twins: would it make sense to name them both “Tim,” on the grounds that it would “level the competitive playing field” as they grow and master their fate? Or do the boys have the right — and the rest of world an interest — in being able to tell them apart? And while we’re on the subject of names, let’s remember that the name “biosimilar” was coined for a reason — as with fraternal twins, who are not identical. (Even if they were — and you wanted to marry one of them — you’d probably want your “choice” to be “informed” by their having distinguishable names.)
In the FTC’s eyes, it seems to come down to clarity of information versus eking out every possible cent of cost savings. It’s not worth the trade-off. Biosimilars have the potential to provide quality alternative medicines and to improve prices in the biologics space. Because of the complexities associated with all biologics (including biosimilars), however, cost savings from biosimilars are not expected to exceed 10 percent to 20 percent over branded products. Chemical compound generics can realize savings of up to 80 percent over brands.
If we go in the direction of non-unique names, and issues arise, we might not have the information we need to quickly understand which among similar products is causing the issue. That can unnecessarily affect trust across a class of drugs and biosimilars as a whole, and that could significantly affect uptake.
Biosimilars are already available in other parts of the world. This gives us a unique opportunity to learn from the experiences of those markets. In Europe, where biosimilars share the same names as the originator product, they’re experiencing an increased number of adverse events, and it can take months for manufacturers to determine if their product is causing the problem.
Thailand also uses nondistinguishable names and rapidly approved biosimilars to treat certain diseases, which has led to both a dramatic increase in the number of cases of life-threatening blood-related adverse events and near futility in efforts to track back to which products are causing the problems. Australia opted for distinguishable codes for all biologics, and they appear to be experiencing successful rollout and uptake of biosimilars.
It’s a universal reality: What’s in a name is a fundamental ability to tell things apart. Nothing more informs American competitiveness and informed consumer choice. No one more than the FTC should recognize that fact — and be its champion.
Peter J. Pitts, a former FDA associate commissioner, is president and co-founder of the Center for Medicine in the Public Interest.
Read More & Comment...According to the FDA Law Blog:
There’s a hot debate brewing over whether or not a biosimilar biological product licensed under Section 351(k) of the Public Health Service Act (“PHS Act”), as added by the Biologics Price Competition and Innovation Act of 2009 (“BPCIA”), should share the same non-proprietary name – or International Nonproprietary Name (“INN”) – as its brand-name reference product counterpart. Last Friday, Amgen Inc. (“Amgen”) further turned up the heat when the company announced the submission of extensive comments (89 pages in length) to two citizen petitions submitted to FDA earlier this year requesting that the Agency require that a biosimilar be identified by the same INN as the reference product it relies on for approval.
The first petition (Docket No. FDA-2013-P-1153) was submitted by the Generic Pharmaceutical Association (“GPhA”) in September and requests that FDA “implement its INN naming policy equally to all biologics” such that all biosimilars “share the same INN name as the” reference product (see our previous post here), The second petition (Docket No. FDA-2013-P-1398) was submitted in October by the Novartis Group of companies (“Novartis”) and requests that FDA “require that a biosimilar[] be identified by the same [INN] . . . as the reference product.” (see our previous post here).
According to Amgen:
Contrary to the position of GPhA and Novartis . . . , we believe that a policy of identical non-proprietary names would pose significant public health risks, and is inconsistent with applicable legal requirements. Petitioners fail to take into account fundamental scientific principles of biological products as well as their distinctive posology and methods of administration in clinical practice. We believe that, both as a matter of public health and as a matter of law, biological products licensed under the BPCIA should have distinguishable non-proprietary names—that is, non-proprietary names comprised of common roots and distinguishable prefixes or suffixes, as deemed appropriate by FDA, to achieve the goals of patient safety through effective post-market surveillance, and widespread physician and patient acceptance and appropriate use of this important new class of medicines.
Amgen lays out its case for distinguishable non-proprietary names – and opposition to the GPhA and Novartis petitions – in seven parts, arguing along the way that:
(1) “Biosimilars licensed under the BPCIA are appropriately neither required nor expected to be structurally identical either to their reference counterparts or to each other. Indeed, the product quality attributes for biosimilars may fall outside even the range of variability that is acceptable for the reference product, and differences in both structure and function will become increasingly prevalent as the complexity of the reference products increases. Because biosimilars cannot be—and are not—brought to market through the same abbreviated pathway as generic drugs, the legal requirements applicable to generic drugs (including the requirement that generics bear the same labeling and established names as their reference counterparts) do not apply to biosimilars. This difference in regulatory treatment is both appropriate and critically important to development of biosimilar medicines using state of the art technology.”
(2) “Distinct safety and immunogenicity profiles are found among structurally related biological products. Similar biological products cannot be fully characterized in premarket clinical trials, but instead must be subject to effective post-market surveillance that distinguishes accurately among related products. . . . [I]t is especially important that pharmacovigilance measures account for the possibility of differences in the safety profiles of related biological products. These differences could be intrinsic to products as a result of their distinct methods of manufacture, or they could be emergent as a result of post-approval manufacturing changes.”
(3) “Because FDA’s spontaneous reporting/post-market surveillance system does not encompass the separate tracking of [products] sharing the same non-proprietary name to be separately tracked, biosimilars with the same non-proprietary names but potentially different immunogenic profiles will be difficult to distinguish, hampering immunogenicity tracking and optimal pharmacovigilance.”
(4) GPhA and Novartis include in their petitions “numerous unsupported and erroneous objections to the use of distinguishable names for biological products.”
(5) A biosimilars naming policy that would mirror the naming rules for generic drugs “would create a significant risk of confusion regarding a biosimilar product’s bioequivalence or identity to its reference product. Moreover, given FDA’s obligations under the Administrative Procedure Act to treat like cases alike and to explain any departures from precedent, FDA would need to reconcile any naming policy with its past statements recognizing the potential safety risks of using identical established names for similar biological products, as well as instances in which FDA has required, for safety reasons, the use of distinguishable names for similar products.”
(6) “[T]the need for better pharmacovigilance measures for all drug products does not obviate the need for distinguishable names to facilitate post-market risk management in the context of biosimilars.”
(7) Alternative pharmacovigilance mechanisms, such as those suggested by GPhA in its citizen petition, “are not sufficient to address the need for robust post-market surveillance of biological products.” Efforts to create a federal tracing system “is not capable of ensuring the robust pharmacovigilance systems necessary for biological products if biosimilars are assigned identical non-proprietary names to their respective reference products.” Read More & Comment...A short missive from White Oak that may portend broader consequences:
“Rachel Sherman has decided to retire after many years of service to both FDA and CDER. We will miss her leadership and guidance as the OMP Office Director and wish her well in future endeavors!”
Wow.
Very few words to announce a very big loss.
Read More & Comment...While Medicare as a whole is a fiscal basket case -- due to run out of money in 2024 -- Part D has been the very model of a well functioning federal program since its implementation in 2006.
The Congressional Budget Office (CBO) found that, between 2004 and 2013, Part D will cost an extraordinary 45 percent below what was initially estimated. Premiums for the program, meanwhile, are roughly half of the government’s original projections. Part D enrollees pay, on average, $30 a month -- a rate that has remained essentially unchanged for years. It’s no wonder that beneficiaries are so pleased with the program. In fact, 96 percent of those enrolled in Part D say that their coverage works well.
These unprecedented results are largely due to Part D’s market-based structure. Beneficiaries are free to choose from a slate of private drug coverage plans, forcing insurers to compete to offer the best options to American seniors. It’s hardly surprising that the program has led to low prices and satisfied customers.
Through their own negotiations with drugmakers, private insurance plans that operate under Part D have already had great success in keeping pharmaceutical prices down. In fact, the CBO has observed that Part D plans have “secured rebates somewhat larger than the average rebates observed in commercial health plans.”
What’s more, the CBO has said time and again that doing away with the non-interference clause “would have a negligible effect on federal spending.” In a report from 2009, they reiterated this view, explaining that such a reform would “have little, if any, effect on [drug] prices.”
Smart partnerships between government and the free market work. They work at keeping costs low and – most importantly – improving care. As JAMA reported, “Implementation of Medicare Part D was followed by increased use of prescription medications, reduced out-of-pocket costs, and improved medication adherence.” And this, in no small measure, significantly reduces more drastic medical interventions -- which in turn reduces our overall national health care spending.
A new report from the Manhattan Institute, “A Decade of Success: How Competition Drives Savings in Medicare Part D,” details many of the reasons why a free-market approach to healthcare access is not only working – but a model for how to design future healthcare partnerships between Uncle Sam and the private sector.
Here is the report’s executive summary:
National trends are not a sufficient explanation for Part D’s success.
While patent expirations are part of the story—national drug spending as a whole slowed during the period we examine—they are far from the full explanation for large overestimates in Part D spending (indeed, patent expirations were likely captured in the original projections). Instead, the available evidence indicates that private-sector firm-level innovations, including preferred pharmacy networks and aggressive negotiations with drug manufacturers, have played a significant role in keeping the program’s costs below projections. We find that broader market trends (e.g., patent expirations and other changes) account for only about half (56 percent) of the program’s performance. The remainder—44 percent—of Part D’s lower-than-estimated cost savings is attributable to factors not captured in national prescription drug trends, which should include competition between Prescription Drug Plans (PDPs). This is strong evidence indicating that consumer-driven competition in Part D has been critical to the program’s financial success.
Consumer-driven competition is a relatively new tool in the government’s effort to control health-care costs.
In hindsight, government overestimates of Part D’s costs are not surprising, since the program utilizes a model of consumer choice (robust competition among dozens of regional drug plans and Medicare Advantage plans) that has no perfect analogue in other government health plans, such as Medicaid.
Part D is an excellent model for future health-care and entitlement reforms.
Arguably, Part D and Medicare Advantage plans represent the first national health-care exchange (the Federal Employees’ Health Benefits Program [FEHBP] is a close cousin). While the Affordable Care Act (ACA) operates a similar exchange concept, there are important differences. First, Part D plans compete in large regional areas, not states (this creates much bigger risk pools because even large states are incorporated into larger regions), as the ACA exchanges do. Even the federal exchange is layered on top of a state-regulated insurance market. This potentially limits the ability of plans to create economies of scale to bargain with providers and to utilize innovative tools to arbitrage cost and quality differences across state markets (preferred pharmacy and mail-order networks in Part D; telemedicine and medical tourism to “centers of excellence” for health-insurance plans). Notably, while Part D includes higher subsidies for sicker seniors (typically, the low-income subsidy population), it does not penalize healthier seniors through higher premiums, as the ACA’s community rating provisions do. Arguably, a better approach would be to rely more on backdoor (non-cross-subsidized) risk adjustment of plans and larger subsidies for sicker or older patients, while allowing plans to charge actuarially fair premiums to younger enrollees. The cross-subsidies in the Part D approach are more transparent in that sense, since they come from tax revenues rather than from private premiums.
The complete MI report can be found here.
Read More & Comment...Per a new draft guidance:
While generic formulations of these drug products are required to be both pharmaceutically and therapeutically equivalent to a reference listed drug (RLD), we are concerned that differences in physical characteristics (e.g., size and shape of the tablet or capsule) may affect patient compliance and acceptability of medication regimens or could lead to medication errors. We believe these patient safety concerns are important, and we are recommending that generic drug manufacturers consider physical attributes when they develop quality target product profiles (QTPPs) for their generic product candidates.
This is important and even more significant considering the agency’s recent proposed rule that would permit generic drug makers to update their labels if they received information about potential safety problems.
(For more on this, see here.)
Next up: tighter ranges for bioequivalency Read More & Comment...In Friday’s Washington Post, the editorial page opines that, “… not every problem can or should be solved by federal regulation, and there is still a decent chance that the balance between regulation and liberty that the Affordable Care Act struck will work. If it does, the more rational health-care system that results will have been well worth the price in expanded government intervention in the health-care market.”
That’s the conclusion, but the body of the piece reads like an introduction to a manifesto about why what we really need is a single-payer system. A "rational health-care system" is the the global code phrase for "one-size-fits-all" care. Have a look here and judge for yourself.
Here’s my view on some of the same problems from a different angle (as seen in yesterday’s Des Moines Register).
The devil is in the details.
Read More & Comment...From: A Message from the Commissioner
Sent: Wednesday, December 11, 2013 02:05 PM
To: FDA-Wide
Subject: Personnel Announcement
Dear Colleagues:
I am deeply saddened to announce that John M. Taylor, Counselor to the Commissioner and Acting Deputy Commissioner for Global Regulatory Operations and Policy (GO), will be leaving the Food and Drug Administration in January 2014 to pursue other opportunities. John has accomplished so much during his two stints at the Agency (from 1991 to 2005 and from 2009 to present) as he served in various ways and touched many lives. From his early days in the Office of Chief Counsel, to his senior roles in the Center for Drug Evaluation and Research, the Office of Regulatory Affairs, and the Office of the Commissioner, John has brought leadership, sound advice, and most of all, good humor to his work. We will all miss his expert knowledge, his innovative thinking, and his uncanny ability to remain calm during times of adversity and matters of urgency. John has proven himself as a leader who epitomizes integrity, dignity, kindness and compassion, and he is widely respected by his colleagues.
Most recently, John has been wearing two hats – Counselor to the Commissioner and Acting Deputy Commissioner for GO. In these two positions over the past year, John has demonstrated flexibility, creativity, and decisiveness that has enabled us to make important forward progress on key matters. John has always stepped up in the times when I have needed him most to take on specific tasks that represent my highest priorities, including those related to globalization, FDASIA, and pharmacy compounding. John has been a catalyst for organizational progress and change while handling and managing the high demands of the Office of the Commissioner. And notably, John agreed to serve as the Acting Principal Deputy Commissioner during a time when the Agency was in a period of transition, for which I am extremely grateful.
I know I speak for all of us when I say that John will be sorely missed. Please join me in thanking him for his distinguished service to FDA and wishing him the very best in his future endeavors.
Margaret A. Hamburg, M.D.
Commissioner of Food and Drugs
In “Personalized Medicine and Responsible Access to Pain Medication” (a white paper based on the Center for Medicine in the Public Interest’s September 2013 Capital Hill conference), Dr. Douglas Throckmorton, CDER’s Deputy Director, for Regulatory Programs and the FDA’s point person on opioids, writes,
We understand that for the millions of Americans experiencing an acute medical need or living with chronic pain, opioids, when prescribed appropriately, can allow patients to manage their pain as well as significantly improve their quality of life. However, we have also become increasingly concerned about the abuse and misuse of opioids. We are challenged with determining how to best balance the need to ensure continued access to patients who need these medications while addressing concerns about abuse and misuse.
FDA must walk a difficult public health tightrope, balancing patient need, medication safety, and (in the case of opioids), the dangers of abuse.
This careful balance is now being called into question by 28 state attorneys general who, in a letter to FDA Commissioner Margaret Hamburg, ask the agency to “reconsider its controversial approval of the powerful new narcotic painkiller known as Zohydro.” The attorneys general are concerned that the medicine lacks “an abuse-limiting formula.”
First let’s consider the rhetoric. Was the approval “controversial?” Well, for starters, the FDA approved the medicine notwithstanding a negative adcomm vote. Is that what makes it “controversial?” No. Adcomm votes are recommendations and the advice (while generally followed by the agency) is in no way binding and there’s plenty of precedent for the FDA going its own way.
No, it’s controversial because the issue of opioid abuse is controversial. And that’s an important difference. Nobody said the FDA’s job was easy.
Noble Prize winner Joshua Lederberg once observed that the failure of regulatory legal and political institutions to integrate scientific advances into risk selection and assessment was the most important barrier to innovation in public health. Lederberg noted that in the absence of such changes, “The precedents affecting the long-term rationale of social policy will be set not on the basis of well-debated principles, but on the accidents of the first advertised examples.” And there isn’t a better perspective-setting proposition when it comes to the issue of Zohydro than that quotation.
Policies and regulations that seek to limit risk are often shaped by the immediate fear of sensational events. This perspective is commonly referred to as the Precautionary Principle, which, in various forms asserts that unless innovators can demonstrate that a new technology is risk free, it should not be allowed into the marketplace. Moreover, any product that could possibly be dangerous at any level should be strictly and severely regulated. But precaution is not always safer than the alternatives.
Pierre Trudeau once said, “There’s no place for the state in the bedrooms of the nation.” But what’s the appropriate place for the state in our nation’s pharmacies and medicine chests? The AGs who are petitioning the FDA see abuse and seek to minimize access to opioids as the solution.
That’s a law enforcement solution. They mean well, but are behaving like a bull in a china shop. Arbitrarily limiting choice is not generally associated with the scientific method. Should regulation be shaped by factors other than science? Or should advances in medicine and digital information be used to right size regulation reduce the excessive reductionism that leads to regulatory overreaction and promote resilience rather than ever increasing regulation.
Which brings us to the second objection raised by the “Opioid-28,” that Zohydro lacks “an abuse-limiting formula.”
Per Doug Throckmorton,
Another important step towards the goal of creating safer opioids, and one that is a high public health priority for FDA is to encourage the development of formulations of these drugs that deter their abuse. This relatively new science of abuse deterrence is exciting and evolving and showing encouraging promise. To guide drug development in this new field, we also issued a draft guidance for industry in January, announcing a flexible, adaptive approach to encourage the development of abuse-deterrent opioids. We believe abuse deterrent products have promise to help reduce prescription drug abuse and improve public health.
The FDA has its eye on the prize. But what’s the game for the AGs? They refer to an “epidemic.” That’s a powerful word. But does it qualify in the case of opioids?
According to the CDC in 2008, there were 14,800 opioid overdose deaths. Half of those, the CDC has claimed, involved opioids and other illicit substances, whether it’s cocaine or heroin, or alcohol. They also mentioned that alcohol was involved in many of those deaths but they don’t actually tell us the numbers. So conservatively, half or 7,400 deaths occurred in 2008 from opioid overdose. The same year from CDC’s own statistics, there were 36,500 suicides. There also were 24,000 alcohol-induced deaths and that doesn’t count other related alcohol deaths like drunk driving. The bottom line is that the opioid numbers do not even come up in the CDC’s list of the top 15 causes of death of Americans
It’s important to add to this “epidemic” perspective, the fact that people suffering from chronic pain are under-served by existing therapies. A recent IOM report that was issued in June of 2011 found that 100 million Americans are now living with chronic pain. That’s a third of the U.S. population. Ten million of those have pain so severe that they are disabled by the pain. The report also said that pain costs the U.S. economy about 600 billion dollars a year in lost productivity and healthcare cost.
And the “Opioid 28” wants fewer pain medications on the market?
The vast majority of people who use opioids do so legally and safely. A subset, approximately four percent, use these medications illegally. In fact, from 2010 to 2011, the number of Americans misusing and abusing opioid medications declined from 4.6% to 4.2%.
And the FDA’s decision was “controversial?” Really?
Rather than dealing with the problem of abuse with sledgehammer solutions (such as those proposed by the 28 AGs), we should focus on potential solutions such as:
* The structure and impact of programs such as the recently instituted by CVS initiative (detailed in a recent New England Journal of Medicine perspective piece) where, through the use of “Big Data”, the chain pharmacy identified outlier prescribers and took appropriate and responsible action.
* The role of the 21st century pharmacist in improving drug safety and medication adherence via more proactive and remunerated patient education? How can pharmacists become better integrated beyond Med Guides into the FDA’s Safe Use of Medicines initiative? When will pharmacy synchronization really kick into gear, and how will states help to jump-start these important initiatives?
* Government and legislative initiatives such as the Stop Act (H.R. 486), which focuses on tamper-deterrent formulations and the continued development of those. Also, Senate Bill 1277 (sponsored by Senator Barbara Boxer, D/CA) which would establish a commission to bring all of the stakeholders together to have discussions about how to approach this issue so that law enforcement, providers, patients, and pharma can debate the issues and reach common ground.
* The appropriate role of tamper-resistant technologies. They are part of the solution, but they’re not the whole solution. We need to develop policy options that focus on the prescriber/patient relationship, and a professional assessment of what’s the risk involving this patient. Is the patient is going to tamper with the medication and potentially expose themselves or others to some danger. We have to do a better job (via CME and other methods) of training physicians and other prescribers on how to do these kinds of assessments.
And, most importantly, we need to keep the needs of patients front and center.
There is no expedient to which a man will not go to avoid the labor of thinking.
– Thomas Edison
As we inch closer to biosimilars in the US, we need to remember that similar does not mean identical – and that means paying close attention to bioequivalence.
MedPage Today reports that FDA-funded studies are underway that may lead the FDA to stiffen its bioequivalence rules for generic anti-epileptic drugs (AEDs) and others with so-called narrow therapeutic indices.
These studies examine drug pharmacokinetics in epilepsy patients and under chronic dosing -- could show that some current generic AEDs vary enough to put patients at risk. My eldest son has Juvenile Myoclonic Epilepsy (JME), and I can attest to the problems with therapeutic substitution from personal experience.
The FDA has shown a willingness to modify its rules if, indeed, the evidence is there, said Barry Gidal, PharmD, of the University of Wisconsin, at a press briefing held at the American Epilepsy Society (AES) annual meeting. "We're bringing the evidence."
Current bioequivalence regulations allow some variation in generic drug pharmacokinetics relative to the original branded drug. In particular, the lower boundary for 90% confidence intervals in measures of bioavailability can be as low as 80% of the mean for the branded drug, and the upper boundary can be as high as 125% of the branded drug mean.
Under current FDA rules, single-dose studies in healthy volunteers are adequate to show bioequivalence. He said that this poses two problems: drug availability and metabolism may be different in patients than in healthy people, and it may also be different with chronic dosing.
The issue of real-world generic equivalence is not confined to AEDs. Immunosuppressants are another drug class with narrow therapeutic indices, and an FDA-funded study of tacrolimus bioequivalence is also underway.
Another view of the bioequivalence issue was provided by another University of Cincinnati study led by Lisa Garrity, PharmD -- a survey of Cincinnati-area pharmacists about their knowledge and experience with generic AEDs.
Garrity and colleagues obtained responses to a one-page questionnaire from 30 retail pharmacists who had a mean of 15.6 years in practice (SD 8.8). Of these, 20 reported having no specific education about possible issues with generic AEDs when switching from one manufacturer to another.
Responses indicated that 22 believed that switching from a branded version to a generic could cause problems, but only 18 said that issues could arise when switching between generic versions of the same drug. Per Garrity, "I think these [types of switches] should be equally concerning.”
Another problem with the current system is the lack of transparency regarding generic drugs' pharmacokinetics. Data from manufacturers' FDA submissions are not readily available.
Can you say INN?
Read More & Comment...Today's FTC hearing on biosimilars cancelled because of safety concerns (snow day in DC).
Read More & Comment...
A good name is better than precious ointment.
-- Ecclesiastes vii. 1.
U.S. approval of biosimilars promise to be a very good thing, but the devil is on the details. Unfortunately, we’re seeing a disturbing trend relating to one of those key details – naming nomenclature.
Advocates for nonproprietary names across innovator reference product biologics and the biosimilars associated with them dangerously miss the mark on the pivotal issues relating to naming. A new editorial in Nature Biotechnology demonstrates such misguided thinking. Wither their usual good sense?
Obviously much education remains to be done on this issue in the time between now and when FDA issues final draft guidance on naming. Because where FDA winds up on this issue -- nonproprietary names, nonproprietary names + identifier codes, unique names or somewhere in between – will significantly impact patients, providers, manufacturers, pharmacists, safety experts and others. We need to all side firmly with what's best for patients.
If you’re for patient safety, you can’t be against distinguishable naming. The WHO established the International Nonproprietary Names (INN) system in 1953 before biologics were a figment of anyone’s imagination. Through the INN system, innovators and generics that share the same active ingredient also share the same generic name, also called the INN. It’s worked pretty well for chemical compounds but, as has been acknowledged by WHO and regulatory bodies of every developed nation, biologics are not chemical compounds – they’re infinitely more complicated.
We need to learn from these market-based experiences of nonproprietary names in the EU and Thailand, and distinguishable names in places like Japan and Australia. We can also take valuable lessons from how approaches specific to naming of biologics lend themselves to more effective safety monitoring, pharmacovigilence, data collection, clarity and transparency.
While the U.S. National Drug Code system will continue to serve a purpose for both small and large molecules, we can’t count on it to be the be-all-end-all solution for safety monitoring for biologics. Not even close. Payers don’t universally use NDC codes, they are rarely present in patient records and they are often inaccurately entered when they are. Distinguishable names provide a necessary safeguard to maximize safety and credibility. It’s really that simple.
The FTC is holding a hearing on the topic of biosimilar naming on Tuesday. They have stacked the deck (with exceptions) and no one is expecting anything other than the susual cost-centric care-verse-patient-safety drivel. I’ll be there all the same trying (from the audience) to interject occasional bouts of patient-centric sanity.
When it comes to biosimilars, we need to be extremely thoughtful about how we set policy relating to these promising medicines and strike a balance that promotes health and safety, rather than forcing a binary response that is driven by profits rather than patients.
Here’s a non-biosimilar quote (with apologies to Mr. Shakespeare):
He that filches from me my good nameRobs me, enriches him,
And makes patients poor indeed Read More & Comment...
Pfizer has announced an update of its clinical trial data access policy that will “simplify and broaden access to information gathered in Pfizer-sponsored clinical trials. The updated policy builds upon and expands the company’s established methods of clinical trial information sharing, including Pfizer’s long track record of submitting for publication results from all interventional clinical trials in patients and its pioneering efforts to provide clinical trial results and data to study participants.”
Key elements of Pfizer’s expanded policy (effective January 1, 2014) are:
* Pfizer’s INSPIIRE public web portal for investigator-initiated research (iirsubmission.pfizer.com) will offer qualified researchers a standard form and process for requesting access to anonymized patient-level data from Pfizer-sponsored trials of approved (or discontinued) products/indications posted on clinicaltrials.gov that have been complete for 24 months.
* An external Independent Review Panel will consider all requests denied or only partially approved by Pfizer and make a final decision.
* Pfizer will publish (on Pfizer.com) synopses of clinical study reports (CSRs) filed with regulatory agencies for approved products for which basic results are posted in the clinicaltrials.gov registry (dating back to September 2007). These CSR synopses will include summary results for all primary and secondary endpoints; any data that could be used to identify individual patients will be removed.
* Pfizer will produce and distribute lay-language summaries of clinical trial results to trial participants who wish to receive them, starting with trials that begin enrolling in 2014, in countries where regulations permit.
* Pfizer is piloting the use of “Blue Button™” technology (launched by the U.S. Departments of Veterans Affairs and Health and Human Services) to enable Pfizer trial participants to download their own electronic clinical data collected in the trial.
According to Pfizer, their expanded clinical data access policy “also reinforces the company’s current practice of submitting for publication manuscripts for all interventional clinical trials in patients, regardless of outcomes, within 18 months of study completion, and the company’s commitment to register and post summary results for interventional human clinical trials to clinicaltrials.gov in the United States and to registries outside the United States as required.”
The full version of Pfizer’s expanded policy, the clinical data request form, the searchable CSR database, the Independent Review Panel membership roster and charter, and more information, including Frequently Asked Questions, are available at http://www.pfizer.com/TrialDataandResults.
Read More & Comment...According to the American Medical Association, cost estimates of inefficient health care claims processing, payment and reconciliation are between $21 and $210 billion. In the physician practice, the claims management revenue cycle consumes an unsustainable 10–14 percent of practice revenue.
The current system is all too often manual. It must be replaced by automated, transparent, unambiguous, real-time health care transactions.
The full AMA white paper on this issue can be found here.
A new article by Robert Oscar, RPh,President and CEO of RxEOB, furthers that argument. According to Oscar:
Prior authorization (PA) is a complex process that is often daunting and monotonous for medical practice managers. It is also costly in terms of economics and human life, in particular when PA requirements lead to patient medication needs “falling through the cracks,” as some patients abandon their prescriptions due to the confusion and delay of the approval process.
Some other important points from Oscar:
The original goal of PA was to save money, requiring physicians to justify to health plans the need for medications, diagnostic tests and procedures, but it has led to pharmacists having to spend an average of five hours per week handling PA requests. This is non-reimbursable time that is better served on direct patient care.
A nationwide physician survey indicates that more than 69 percent of physicians typically wait several days to receive a PA from an insurer for a prescribed drug, while 10 percent wait more than a week. While more than 52 percent of office-based prescribers utilize electronic prescribing methods, most of them continue to use paper-based methods for obtaining PA of medications from health plans, causing unnecessary delays for patients.
The good news is that a growing number of physicians and office managers are taking proactive steps to solve these issues by taking advantage of electronic PA (ePA), a technology that enables them to submit an authorization request through hand-held devices or via a web portal prior to pharmacy adjudication.
ePA speeds up health insurer response time, minimizes resources associated with manual processes, and helps to enhance the quality and utility of the PA process.
For physicians and office practice managers who want to reduce manual PA workflow, improve the quality of the PA enforcement process, automatically document all activities, reduce the PA approval response time, and increase their understanding of lower-cost therapeutic alternatives, ePA technology is the way to go. The best solution can enable physicians and their staff assistants to submit the PA request online via EMR or e-prescribing workflow, thereby mitigating unnecessary delays, improving the quality of patient care and enhancing the patient’s overall experience with care delivery process.
Oscar’s complete analysis can be found here.
Read More & Comment...Yesterday I participated in a conference on Content Marketing. (The complete program agenda can be found here.)
Content marketing?
As my sister (a mental health professional) asked me, “Content marketing? As opposed to what, bullshit marketing?”
Not a question I was prepared for over the Thanksgiving table but, from someone who we generically refer to as “a provider,” an honest and relevant one.
Rhetoric counts. Maybe a better phrase is “content management” – because then we can compare it to something more tangible – such as “financial management.” We certainly know what that is. You take a certain about of money (the “content”) and through a savvy understanding of the marketplace and using the legal tools and compliant instruments you seek to increase the value of your portfolio.
When it comes to healthcare, is it content marketing or content management – or is it yet something else?
Maybe a better way to ask the question is, if it’s content marketing, what is the content and to whom are we marketing it – and why?
Content marketing, as a business proposition, is about maximizing awareness, reputation, sales, market share – and the advancement of the public health. (And it needn’t be in that order.) Indeed, the purpose of content marketing is to maximize the potential of important, accurate and timely information.
So maybe we should be talking about content maximization – and a more three-dimensional agenda – that is to say, beyond sales acquisition to driving patient outcomes.
(Another reason to alter the nomenclature is to help recalibrate the corporate compass.)
Some important questions:
How can content maximization address the adherence/compliance quandary?
How can content maximization help healthcare communicators advance the use of new platforms and media?
Most importantly, how can content maximization help define healthcare communications in a post-blockbuster environment, specifically as it pertains to orphan diseases and the rise of personalized medicine?
Let’s consider how the goal of content maximization through the strategy of content management and the tactics of content marketing can help advance sales, corporate, and public health goals. (And, again, not necessarily in that sequence.)
The heart of content marketing is story telling. Savvy healthcare marketers need to move from ABC (“Always Be Closing”) to ABT (Always Be Telling).
In the world of 21st Century healthcare, companies must share their content from their own mouths – because in the ultra-transparent world of social media, you can't separate the story from the storyteller. And we shouldn't even want to.
Rhetoric counts. As Kurt Vonnegut wrote, we need to “transcend the bullshit.”
Amen.
Read More & Comment...It’s fast becoming an n of 1 world. A world where every disease is an orphan disease and success is measured by individual outcomes rather than large population studies such as CATIE or ALLHAT or the multitude of programs being funded by PCORI.
Small is the new Big means that we must also think differently about pharmacovigilance. While we must continue to capture adverse event data (and do a better job at that through social media channels and mobile apps), we must also strive to capture Substandard Pharmaceutical Events (SPEs). SPEs occur when a pharmaceutical product does not perform as expected – perhaps because of API or excipient issues. SPEs can arise because of an issue related to therapeutic interchangeability. When it comes to 21st century pharmacovigilance, we have to both broaden and narrow our views about bioequivalence to the patient level. Small is the new Big.
And when it comes to drug development, adaptive clinical trials and companion diagnostics further define the outsized urgency of small-scale thinking. Demonstrating outcomes on an n of 1 level is crucial not just for 21st century healthcare technology assessment (think “value-based reimbursement”) but also for physician pay-for-performance measures and, last but not least, for the benefit of actual patients
There’s a lot of lip service paid to the comment that “the era of the blockbuster is over.” Now consider that statement from the perspective of another industry – in the 21st Century would you rather be Blockbuster or Netflix?
Small is the new Big. And that means a focus on individual patient outcomes, which means a focus on the individual patient rather than the general population and on long term care rather than short term cost.
And it’s about time. Read More & Comment...CDER Staff:
Today, FDA announced it will require the removal of certain prescribing and use restrictions for the diabetes drug, Avandia (rosiglitazone), to reflect new safety information regarding Avandia’s cardiovascular risk. The changes include modifications to the drug label about cardiovascular safety, changes to the Risk Evaluation and Mitigation Strategy (REMS), and the removal of a requirement for the drug’s maker to do another study of the drug.
In 2007, concerns had been raised about an elevated risk of heart attacks and strokes, and related deaths associated with Avandia. FDA issued safety recommendations to the health care community and initiated an intensive look into the cardiovascular risk of the drug. In 2010, GlaxoSmithKline (GSK) released a long-term study called the Rosiglitazone Evaluated for Cardiovascular Outcomes and Regulation of Glycemia in Diabetes (RECORD) trial, which evaluated whether patients are at greater risk of heart attack or death when taking Avandia, as compared to standard-of-care diabetes drugs. As a result of this study, GSK said it could find no potential elevated risk.
Questions continued to be raised both internally and externally about Avandia’s safety and the RECORD study. In light of the inconclusive scientific evidence, I made the decision that Avandia could remain on the market but with restricted access, until we had further data about the cardiovascular risk of this drug. An independent analysis, or readjudication, of the RECORD trial was conducted by the Duke Clinical Research Institute (DCRI) and confirmed the original findings by GSK, lending stronger evidence about the safety profile for Avandia. In June of this year, we convened two advisory committee panels to discuss the results of the DCRI review of the RECORD trial. The majority of the advisory committee panelists concurred with the findings and voted in favor of easing the 2010 restrictions on the drug. Our action today is consistent with the advice of those expert panelists.
I believe the Center’s work on Avandia is noteworthy for at least three reasons, as it:
1) Exemplifies our lifecycle approach to drug regulation, and our ongoing work to monitor the safety and effectiveness of medications once they are on the market, and make necessary adjustments when the risk/benefit balance of a drug changes over time
2) Underscores the complexity of our decision-making processes, and how as regulators, we must make decisions about regulated products -- even in the face of uncertainty or in the absence of complete information about a drug’s safety or effectiveness
3) Reinforces the importance of our commitment to Equal Voice and our efforts to encourage viewpoints from our expert disciplines across CDER, and even when there is disagreement, to respect the views of each discipline and strive to make the best decisions possible based on the scientific evidence available and in the best interest of patients
A wide range of scientific disciplines and many people from a number of CDER offices have been involved in our efforts to characterize and communicate to the public the cardiovascular safety profile for Avandia. These offices include the Office of Surveillance and Epidemiology, Office of New Drugs, Office of Executive Programs’ Division of Advisory Committee and Consultant Management, Office of Communications, and statisticians in the Office of Translational Sciences. I greatly appreciate the work and professionalism of so many of our staff throughout this process and believe it can be instructive going forward in how we approach and attempt to resolve very difficult drug safety issues.
For more information about the changes announced today, visit FDA requires removal of certain restrictions on the diabetes drug AvandiaJanet Woodcock Read More & Comment...
Mom, Apple Pie, and NIH Funding
Is NIH funding really the be-all/end-all of healthcare innovation?
Mari Serebrov, Washington Editor of BioWorld writes, “With the sequestration blade set to indiscriminately shave federal programs again in January, a bipartisan group of senators is urging congressional budget negotiators to spare research dollars at the National Institutes of Health (NIH).”
But is NIH really the only game in town.
Such a singular focus on the basic research funded by NIH ignores the fact that other government agencies are doing important health care research, Peter Pitts, president of the Center for Medicine in the Public Interest, told BioWorld Today. That research also is getting nicked by the sequester razor.”
For instance, the FDA funds research into regulatory science and personalized medicine, despite a “very limited budget,” Pitts said. While regulatory science research may not seem as sexy as basic research into cancer cures, it’s necessary research, he added.
“Basic research is important,” Pitts said, “but it’s not the beginning, middle and end” of all health care research.
The concerns also ignore new government funding for other types of research. Pitts described the current U.S. funding of public health-related research as “a bigger pie with more people with knives and forks.” More research is taking place, but the NIH slice of the pie isn’t getting any larger.
A prime example of that growth was the creation of the Patient-Centered Outcomes Research Institute (PCORI) in 2010. Set up as a nonprofit under the Affordable Care Act, PCORI was charged with funding comparative-effectiveness research. While Congress began shaving NIH funding close to the jaw line, it mandated that $10 million be set aside in fiscal 2010 to fund PCORI’s activities, $50 million in fiscal 2011 and $150 million in fiscal 2012.
To date, PCORI, which is funded through transfers from two Medicare trust funds rather than general tax dollars like the NIH, has approved 197 research awards totaling more than $273.5 million. It also is committing more than $1 billion to research funding over the next two fiscal years. But that money will be used to “study other people’s research, rather than the basic research NIH promotes,” Pitts said.
Noting that all research is subjective, Pitts said it isn’t Congress’ job to approve individual research projects, but Congress makes the choices of how federal research dollars are spent in general.
The complete BioWorld article can be found here.
Read More & Comment...
According to Peggy Hamburg ...
The difference between science and science fiction is a line that seems ever harder to distinguish, thanks in part to a host of astonishing advances in medical science that are helping to create a new age of promise and possibility for patients.
Today cancer drugs are increasingly twinned with a diagnostic device that can determine whether a patient will respond to the drug based on their tumor’s genetic characteristics; medical imaging can be used to identify the best implantable device to treat a specific patient with clogged coronary arteries; and progress in regenerative medicine and stem cell therapy using a patient’s own cells could lead to the replacement or regeneration of their missing or damaged tissues. Given these trends, the future of medicine is rapidly approaching the promising level of care and cure once imagined by Hollywood in futuristic dramas like Star Trek.
But these examples are not science fiction. They are very real achievements that demonstrate the era of “personalized medicine” where advances in the science of drug development, the study of genes and their functions, the availability of increasingly powerful computers and other technologies, combined with our greater understanding of the complexity of disease, makes it possible to tailor treatments to the needs of an individual patient. We now know that patients with similar symptoms may have different diseases with different causes. Individual patients who may appear to have the same disease may respond differently (or not at all) to treatments of that disease.
FDA has been playing a critical role in the growth of this new era for a number of years. Even before I became FDA Commissioner the agency was creating the organizational infrastructure and putting in place the regulatory processes and policies needed to meet the challenges of regulating these complex products and coordinating their review and oversight. It has been my pleasure to serve at FDA during this next exciting period and to help ensure that the agency continues to prioritize this evolution by anticipating, responding to, and encouraging scientific advancements.
I am very pleased to be able to present a new report by FDA as part of our ongoing efforts in this field. Paving the Way for Personalized Medicine: FDA’s Role in a New Era of Medical Product Development describes many of the exciting developments and looming advances in personalized medicine, lays out the historical progress in this field, and examines FDA’s regulatory role: from ensuring the availability of safe and effective diagnostic devices, to addressing the challenges of aligning a drug with a diagnostic device, to post-market surveillance.
Outside collaboration and information sharing is essential for this field to flourish. On Tuesday, the American Association for Cancer Research and AdvaMedDX held a fruitful daylong conversation on personalized medicine to treat cancer. I was one of the speakers, participating in a conversation with Dr. Francis Collins, the head of the National Institutes of Health. Our discussion focused in part on current status of drug and diagnostic co-development and the challenges and potential of whole genome sequencing, where data can be collected on a patient’s entire genetic makeup at a reasonable cost in a reasonable amount of time.
FDA is committed to fostering these cooperative efforts, as it will require the full force of government, private industry, academia and other concerned stakeholders to maximize our efforts and fully realize the promise of personalized medicine. Our new report outlines that commitment, and helps chart the way forward so that more people can live long and prosper.
Read More & Comment...Via the New Republic:
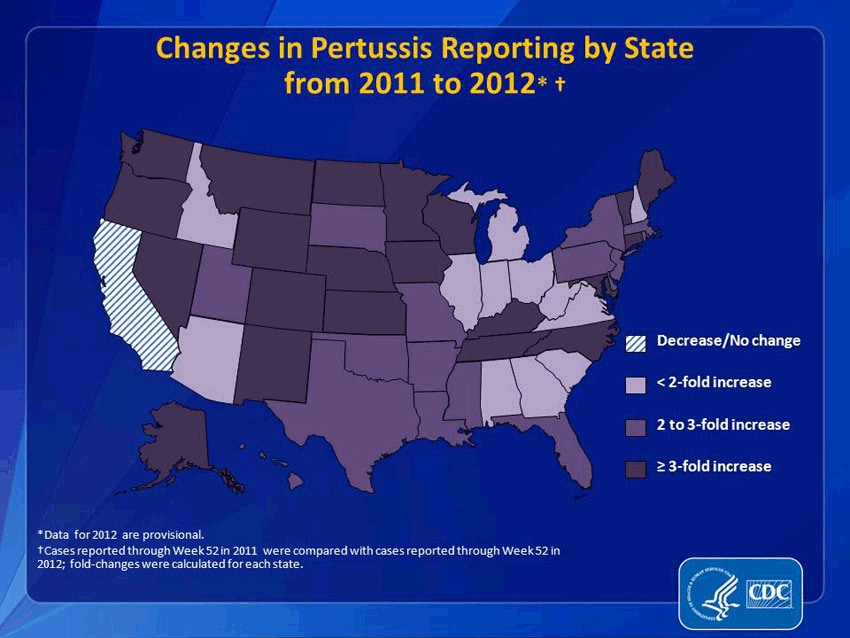
It would be an understatement to say that pertussis and other formerly conquered childhood diseases like measles and mumps are making a resurgence. Pertussis, specifically, has come roaring back. From 2011 to 2012, reported pertussis incidences rose more than threefold in 21 states. (And that’s just reported cases. Since we’re not primed to be on the look-out for it, many people may simply not realize they have it.) In 2012, the CDC said that the number of pertussis cases was higher than at any point in 50 years. That year, Washington state declared an epidemic; this year, Texas did, too. Washington, D.C. has also seen a dramatic increase. This fall, Cincinnati reported a 283 percent increase in pertussis. It’s even gotten to the point that pertussis has become a minor celebrity cause: NASCAR hero Jeff Gordon and Sarah Michelle Gellar are now encouraging people to get vaccinated.
{kind=link}
How responsible are these non-vaccinating parents for my pertussis? Very. A study recently published in the journal Pediatrics indicated that outbreaks of these antediluvian diseases clustered where parents filed non-medical exemptions—that is, where parents decided not to vaccinate their kids because of their personal beliefs. The study found that areas with high concentrations of conscientious objectors were 2.5 times more likely to have an outbreak of pertussis. (To clarify: I was vaccinated against pertussis as a child, but the vaccine wears off by adulthood, which, until recently, was rarely a problem because the disease wasn't running rampant because of people not vaccinating their kids.)
So thanks a lot, anti-vaccine parents. You took an ethical stand against big pharma and the autism your baby was not going to get anyway, and, by doing so, killed some babies and gave me, an otherwise healthy 31-year-old woman, the whooping cough in the year 2013. I understand your wanting to raise your own children as you see fit, science be damned, but you're selfishly jeopardizing more than your own children. Carry your baby around in a sling, feed her organic banana mash while you drink your ethical coffee, fine, but what gives you denialists the right to put my health at risk—to cause me to catch a debilitating, humiliating, and frightening cough that, two months after I finished my last course of antibiotics (how’s that for supporting big pharma?), still makes me convulse several times a day like some kind of tragic nineteenth-century heroine?
If you have an answer, I’ll be here, whooping, while I wait.
Read the full article here.
Read More & Comment...
Social Networks
Please Follow the Drugwonks Blog on Facebook, Twitter, LinkedIn, YouTube & RSS
Add This Blog to my Technorati Favorites