DrugWonks on Twitter
Tweets by @PeterPittsDrugWonks on Facebook
CMPI Videos

Video Montage of Third Annual Odyssey Awards Gala Featuring Governor Mitch Daniels, Montel Williams, Dr. Paul Offit and CMPI president Peter Pitts

Indiana Governor Mitch Daniels

Montel Williams, Emmy Award-Winning Talk Show Host

Paul Offit, M.D., Chief of the Division of Infectious Diseases and the Director of the Vaccine Education Center at the Children’s Hospital of Philadelphia, for Leadership in Transformational Medicine

CMPI president Peter J. Pitts

CMPI Web Video: "Science or Celebrity"
Tabloid Medicine

Check Out CMPI's Book
A Transatlantic Malaise
Edited By: Peter J. Pitts
Download the E-Book Version Here
CMPI Events

Donate

CMPI Reports

Blog Roll
AHRP
Better Health
BigGovHealth
Biotech Blog
BrandweekNRX
CA Medicine man
Cafe Pharma
Campaign for Modern Medicines
Carlat Psychiatry Blog
Clinical Psychology and Psychiatry: A Closer Look
Conservative's Forum
Club For Growth
CNEhealth.org
Diabetes Mine
Disruptive Women
Doctors For Patient Care
Dr. Gov
Drug Channels
DTC Perspectives
eDrugSearch
Envisioning 2.0
EyeOnFDA
FDA Law Blog
Fierce Pharma
fightingdiseases.org
Fresh Air Fund
Furious Seasons
Gooznews
Gel Health News
Hands Off My Health
Health Business Blog
Health Care BS
Health Care for All
Healthy Skepticism
Hooked: Ethics, Medicine, and Pharma
Hugh Hewitt
IgniteBlog
In the Pipeline
In Vivo
Instapundit
Internet Drug News
Jaz'd Healthcare
Jaz'd Pharmaceutical Industry
Jim Edwards' NRx
Kaus Files
KevinMD
Laffer Health Care Report
Little Green Footballs
Med Buzz
Media Research Center
Medrants
More than Medicine
National Review
Neuroethics & Law
Newsbusters
Nurses For Reform
Nurses For Reform Blog
Opinion Journal
Orange Book
PAL
Peter Rost
Pharm Aid
Pharma Blog Review
Pharma Blogsphere
Pharma Marketing Blog
Pharmablogger
Pharmacology Corner
Pharmagossip
Pharmamotion
Pharmalot
Pharmaceutical Business Review
Piper Report
Polipundit
Powerline
Prescription for a Cure
Public Plan Facts
Quackwatch
Real Clear Politics
Remedyhealthcare
Shark Report
Shearlings Got Plowed
StateHouseCall.org
Taking Back America
Terra Sigillata
The Cycle
The Catalyst
The Lonely Conservative
TortsProf
Town Hall
Washington Monthly
World of DTC Marketing
WSJ Health Blog
DrugWonks Blog
In case you missed this story over the holidays.
WASHINGTON (AP) -- The Food and Drug Administration approved 41 first-of-a-kind drugs in 2014, including a record number of medicines for rare diseases, pushing the agency's annual tally of drug approvals to its highest level in 18 years. FDA drug approvals are considered a barometer of industry innovation and the federal government's efficiency in reviewing new therapies. Last year's total was the most since the all-time high of 53 drugs approved in 1996.
The 2014 approval list includes 15 drugs for so-called orphan diseases, which are rare conditions and disorders that affect fewer than 200,000 people in the U.S. Last year's tally, which included drugs for rare cancer and metabolic disorders, exceeded the 13 orphan drugs approved in 2012.
Nine drug approvals in 2014 benefited from the FDA's "breakthrough" designation, a recent program designed to speed up development of promising drugs by providing companies with extra meetings and earlier communication with FDA scientists. Milne said these meetings provide more predictability and transparency about the FDA review process, a boon to both companies and investors.
The complete AP story can be found here.
Read More & Comment...Have a look at this video and please pass it on. Read More & Comment...
That plaque has been tarnished by the unwillingness of Brandeis University to live up to that credo. Rather than immediately defend Brandeis student Daniel Mael’s right to republish a fellow student's call for intifada in America and her lack of remorse over of the death of the two NYPD officers, Brandeis has let radical forces threaten Mael’s safety.
As Lori Lowenthal Marcus wrote: “There’s an ugly tempest brewing at Brandeis University and it’s based, at least in part, on free speech, tolerance and student safety. The storm grew out of a more generalized anger with the state of public discourse and of the safety of individuals in our society at large.
But at this point, one black self-described revolutionary and one Jewish conservative journalist, both Brandeis students, are the figureheads in a battle for the soul of an institution.”
That battle was lost long ago. Brandeis, like many other universities now limit the actions and speech of anyone who differ with those groups who define themselves as oppressed or as defending the oppressed.
Like many other students, Kadijah Lynch is embracing a radical, wholesale attack on America’s democratic capitalism that begins with limiting the freedom of those who disagree with you or dare to criticize you and ends with those that are ‘oppressed’ deciding what the oppressors can do.
To the radical, suppression of free speech is necessary if the society is unjust and unequal. The ruling classes and its apologists will use political freedoms and civil liberties to stay in power. And anyone who disagrees with any aspect of the program or worldview is regarded as a threat to revolution. Hell, freedom is simply a bourgeois prejudice, as Lenin put it.
So attacking Daniel Mael for reposting ugly Tweets from a fellow student and self-styled revolutionary is not wrong, it’s moral.
And Brandeis University – or at least a significant share of the administrators, professors and students feel the same way.
Some would point to the University’s invitation to have Jimmy Carter speak about the evils of Israel and dis-honoring Ali Al-Hirsi because she had spoken about the evils of radical Islam as examples of this allegiance to a radical form of egalitarianism. Indeed, bashing Israel and defending Islam is the most common way to demonstrate support for ‘the oppressed.’ That explains why the anti-police forces have drawn the attention of ISIS, Al Qaeda and why the social media mob attacking Mael is led by the Hamas campus organization, Students for Justice in Palestine
Brandeis not only practices a form of ‘free speech’ that encourages such groups, it is the fountainhead of what Herbert Marcuse – one of its former professors – called repressive tolerance.
Marcuse believed that in America freedom perpetuates injustice. In 1965, while still at Brandeis, he wrote a highly influential article entitled, "Repressive Tolerance“ As a Wikipedia article on Marcuse argues that “genuine tolerance does not permit support for "repression", since doing so ensures that marginalized voices will remain unheard. He characterizes tolerance of repressive speech as "inauthentic." Instead, he advocates a form of tolerance that is intolerant of right wing political movements. That is, the liberation of the oppressed requires silencing those who oppose them. As Marcuse asserts:
“This means that the ways should not be blocked on which a subversive majority could develop, and if they are blocked by organized repression and indoctrination, their reopening may require apparently undemocratic means. They would include the withdrawal of toleration of speech and assembly from groups and movements which promote aggressive policies, armament, chauvinism, discrimination on the grounds of race and religion, or which oppose the extension of public services, social security, medical care, etc.
As to the scope of this tolerance and intolerance: ... it would extend to the stage of action as well as of discussion and propaganda, of deed as well as of word.”
While teaching at Brandeis, Marcuse was afforded every opportunity to speak his mind. True his worldview, Marcuse took every chance to criticize those who disagreed with him. Abraham Sachar, the President of Brandeis who hired Marcuse in 1954 said that Marcuse was “the most relevant symbol in my experience of the cynical radical who demands independence as a right for himself but considers it ‘hypocrisy’ when it is expected of him.” Sachar goes on to note that Marcuse was not fired for his views but rather quit to accept a longer contract from another university. Sachar writes that Marcuse never sought to the set the record straight. Rather, he remained silent.
Today Brandeis has endorsed the ‘narrative’ of a radical professor being fired for his views. It recently celebrated Marcuse with a one-day conference entitled: “The Many Dimensions of Herbert Marcuse.” An ironic title since the event was an intellectual love fest that skated over the discussion of repressive tolerance. The International Herbert Marcuse Society largely organized the event. The group consists of diehard radicals who view the organization as an engine of repressive tolerance. So for instance, the next Society conference focuses on the following question: “ Given Marcuse’s emphasis on praxis, critical pedagogy cannot be limited to classroom space in universities - how can a critical rationality translate into programs of activism, agitation, and organization?” In other words, how can universities be turned into factories of radicalization and repression?
Marcuse dedicated “Repressive Tolerance” to his students at Brandeis including Angela Davis and Abbie Hoffman. Davis became closely associated with the Black Panthers, the Communist party and most notoriously, with providing her sawed-off shotgun to kill a judge who had sent her lover, George Jackson, to prison. She was acquitted and later received the International Lenin Peace Prize (formerly named the International Stalin Peace Prize) by East Germany. These events have been scrubbed from the official Brandeis University biography of Davis.
The failure to defend Daniel Mael’s right to republish the vile and violent comments of Ms. Lynch is the result of Brandeis University’s passive acceptance of repressive tolerance. It is a sad decline.
Sachar recalls that Marcuse and other leftist professors organized a rally at Brandeis during the Cuban Missile Crisis. They called the Khrushchev-Castro decision to based misses in Cuba as a legitimate response to “American imperialism.” One speaker, anthropology professor Katherine Gough, declared to wild applause, that if war broke out she hoped America would be defeated.
When Sachar found out about her comments, her met with her to find out first hand what she said. Hough repeated her statements. Sachar told her that he did not “quarrel with her right to denounce American foreign policy. The freedom of platform gave her no warrant for an irresponsible attack.”
Gough’s husband David Abele (head of the anthropology department) claimed that Sachar’s criticism was illegitimate. Sachar replied “apparently freedom of speech means that you can dish it out but you don’t have to take it.” To which Abele said: when I speak it’s my right. When you speak its intimidation. Incredibly, Abele then requested a bonus for his wife. Sachar said no. Abele and Gough left for another university, but not before suing Brandeis for ‘unfair treatment.’
Sachar did not reward Gough or Abele for their behavior. His determination to uphold the credo “to speak freely, to question openly, to differ without fear” caused those that attacked that principle to leave Brandeis.
Up till now, the Brandeis community and its president, Fred Lawrence, have failed to defend Daniel Mael and speak out against repressive tolerance. Next week, when Mael and his parents meet with Lawrence, who is considered to be the front runner to head the Anti-Defamation League. The president will have a choice: the valor of Abraham Sachar or Marcuse’ radical program of suppression.
Read More & Comment...
Baby, it’s cold outside – if you’re suffering from Hep C.
A recent article in the Washington Post, “One company’s battle to stop the wave of high priced drugs,” could more accurately have been titled, “One company’s battle to stop innovation.”
The Post reports:
This week, the country's biggest drug plan manager Express Scripts said it will drop Gilead's drugs Sovaldi and Harvoni from a list of preferred drugs covering 25 million patients and instead will only put Viekira Pak on its list. Express Scripts said it has negotiated a significant discount with AbbVie — perhaps between $20,000 and $30,000 per course of treatment — in return for expanding access to patients and allowing primary care doctors to prescribe the drug … Until recently, Express Scripts didn't exclude any drugs from its list of preferred medications. But the exclusion list grew from 44 drugs two years ago to 66 in this past year.
Until recently, Express Scripts didn't exclude any drugs from its list of preferred medications. But the exclusion list grew from 44 drugs two years ago to 66 in this past year. The company's chief medical officer, Steven Miller, said he's been telegraphing a move like the AbbVie deal all year.
"We need innovation, and affordability has to be part of that innovation," Miller said in an interview. The companies aren't disclosing the discount, but he said the deal helps close the gap between prices in the U.S. and western Europe. Sovaldi is priced at $57,000 in the United Kingdom and $66,000 in Germany.
That’s the party line – for payers. But you have to read on to get the rest of the story -- about the chilling impact the Express Scripts strategy will have on patient care and pharmaceutical innovation.
"The only way you do exclusions," Miller said, "is you have to do drugs that clinically have identical outcomes."
But Dana Goldman, director of the Schaeffer Center for Health Policy and Economics at the University of Southern California, warns this could come at the expense of the patient since not all will respond to the same treatment. Express Scripts says it won't limit access for hepatitis C patients who need Gilead's drugs, but drug exclusions could still be problematic for patients, Goldman said.
"The reality is if you put up barriers for physicians and patients, we know that people won't have as good access," said Goldman, whose own research found that limits on what drug availability lead to worse outcomes for psychosis patients.
What’s really happening is insurers want someone else to pay for their failure to adequately price demand for highly effective, potentially lifesaving drugs. If the critics had their way and new regulations required price slashing, inevitably patients would lose access to lifesaving therapies, both directly and as a result of reduced research and development expenditures on what could be the next Sovaldi, or Ebola-fighting ZMapp.
Insurers also are hardly powerless, which is evident in their ability to shift drug costs to patients. While critics lambaste the American health system as free enterprise run amok, in reality the U.S. health insurance sector is more like a regulated monopoly – with a mandated customer base that will keep growing as Obamacare expands its reach and as America continues to age.
Prescription drugs currently make up just over 11 percent of the nation’s nearly $3 trillion health care tab; simple math indicates pharmaceuticals are not the major driver of runaway U.S. health expenditures.
And Express Scripts is not what anyone would call a disinterested party.
Consider the 2010 comment of George Paz, chairman and chief executive of Express Scripts, “The cheapest drugs is (sic) where we make our profits.” And just who is “cheaper” better for? "Our whole model is switching people to lower-cost drugs. The more money my shareholders make, the more money I make."
Paz ranked sixth on the 2012 Forbes CEO compensation list, with $51.5 million in total compensation the preceding year, and $100.2 million over a five-year period.
More money for George and Co., but less choice and no savings for patients. This has been the case with brand vs. generic medicines for years. But at least these often resulted in lower out-of-pocket co-pay expenses for patients. Today, the same fatten-George’s Wallet schemes are being used for drugs for evermore serious and life-threatening conditions – such as Hep C. Express Scripts enjoys enormous leverage in the marketplace. What Express Script’s Dr. Miller didn't’ say about drug exclusions, is that his employer told investors earlier in the year that company planned to save $1 billion in 2015 by excluding 66 medicines from its list of covered drugs.
And they did.
Brr. That’s the sound of chilling innovation.
Read More & Comment...One of the myths perpetuated during the debate over the Affordable Care Act (and nowhere more so than in the Michael Moore movie “SiCKO”) was that it would provide “free” health care “like in Europe.”
Such myths die hard – and especially in Vermont.
The Wall Street Journal writes:
Last week, in a reversal that deserves more attention, Democratic Governor Peter Shumlin announced that Vermont would no longer create America’s first statewide single-payer health system. Vermont was seeking a waiver from the Affordable Care Act to abolish what’s left of the nominally private insurance market by 2017, but Mr. Shumlin’s budget gremlins concluded the plan was too expensive and would damage the state economy … The state accountants estimated that his plan required an 11.5% tax on worker payroll, with no exceptions. Individuals, meanwhile, would have paid as much as 9.5% of earnings, which would have applied to everyone making more than four times the poverty level, or $102,220 for a family of four—hardly the 1%. The full $2.59 billion in necessary funding would roughly double current state revenues (about $2.85 billion today).
(The full Wall Street Journal editorial can be found here.)
In other words, there is no such thing as “free” government-supplied health care.
This shouldn’t have come as a surprise. In Canada, while the percentage of taxes used to provide health care varies, it’s estimated that about 22% of taxes collected go into the health system and several provinces, including Quebec, Ontario, Alberta, and British Columbia also charge additional premiums. Citizens in the U.K. pay 11% of each pound they make in weekly income, between £100 and £670 for the NHS,
And what can’t be overlooked is that price controls equals choice controls.
When it comes to the Affordable Care Act, patients can access any medicine they need -- as long as it's on the exchange formulary. Sure, the ACA limits the degree to which insurers can charge higher premiums for sicker patients, but ObamaCare plans found a way around these rules: impose higher out-of-pocket costs for all or most specialty drugs. High co-pays effectively remove choice from the system for many patients.
The breakdown of Silver plans (the most popular category) is particularly revealing. In seven classes of drugs for conditions from cancer to bipolar disorder, more than a fifth of these plans require patients to shoulder 40 percent of the medicine’s cost. And 60 percent of Silver plans place all drugs for illnesses like multiple sclerosis and rheumatoid arthritis in the “formulary tier” with the highest level of cost-sharing.
Nearly every Silver plan across the country, in fact, puts at least one class of drug exclusively in the top cost-sharing tier. In effect, this leaves patients with a given condition — whether HIV or Crohn’s disease — without a single affordable treatment option. Silver is the new Black.
Referring to the Model T, Henry Ford famously said, "Any customer can have a car painted any color that he wants so long as it is black.” That worked out fine – until there was competition. Choice is the great emancipator. The same is true when it comes to healthcare – and a lot more important.
Read More & Comment...Improving the Affordable Care Act to help the chronically ill
By Larry Hausner
December 20, 2014
The Hill
This year, one in three Americans chose to forego medical care for themselves or a family member because of cost concerns, according to a new Gallup poll.
The Affordable Care Act was supposed to prevent those situations and extend reasonably priced health care to everyone. But insurance companies have been exploiting loopholes in the law to avoid this obligation.
Fortunately, the Centers for Medicare and Medicaid Services -- the federal agency that regulates health insurance -- recently proposed a new rule that will close such loopholes and strengthen patient protections.
As the organization finalizes its regulations, it has come under pressure from insurance industry lobbyists to water them down. The CMS should resist these efforts and install genuine reforms that will improve health coverage for vulnerable Americans.
These new regulations take aim at one of the biggest shortcomings of the Affordable Care Act -- unreasonably high patient cost-sharing. The healthcare law caps out-of-pocket expenses at $6,350 for individual plans and $12,700 for family plans. After an insured person or family hits the cap, the insurance company must pay the rest of the treatment costs.
However, insurers have found a way around that cap by forcing families to pay all the way up to that $12,700 threshold even when only a single household member is sick. The new CMS regulations would prohibit insurance companies from charging more than $6,350 for treating any one patient.
This change protects families and furthers the original principle of the ACA: no one should go bankrupt because of disease.
Insurance companies have also been burdening patients who have qualified for so-called "cost-sharing reduction" plans. These plans feature lower out-of-pocket maximums than standard plans, but are available only to patients earning less than 250 percent of the federal poverty line.
Many of these plans include absurdly high co-insurance rates. A new report by Milliman, a major consulting firm, finds that 22 percent of cost-sharing reduction plans feature co-insurance rates of 50 percent or more for certain specialty drugs. In other words, insurance companies are forcing some of the poorest patients in the country to pay the lion's share of their prescription drug costs.
The new CMS regulations will require insurers to summarize the coverage and benefits for these cost-sharing reduction plans and present that information clearly to customers. That's a welcome move, but the agency could go further by prohibiting insurers from charging poor patients co-insurance at all.
The new rule also addresses the problem of rampant insurer discrimination against some of the most vulnerable Americans -- those suffering from serious diseases like diabetes, cancer, and multiple sclerosis.
A study by Avalere Health, a prominent healthcare advisory company, discovered that an alarming 86 percent of the most common insurance plans sold through the new state-level exchanges placed all medications for at least one drug type in the highest cost-sharing tier.
In practice, this means that, say, a diabetes patient might be hit with extremely high out-of-pocket expenses when she goes to fill a prescription for key treatments. The Avalere study found that many exchange plans require 30 percent or more "co-insurance" for all diabetes drugs. Co-insurance forces patients to bear a set percentage of the final drug bill, regardless of how big it is.
In its proposed rule, CMS clarifies that this practice is a form of discrimination against patients. This call-out is commendable but insufficient. The agency should also prohibit companies from pricing all drugs of a particular type out of reach for average patients.
The proposed CMS rule will stop insurers from another method of discrimination -- changing a plan's cost-sharing or benefit structure halfway through the year, after consumers are already locked-in. CMS should also make sure insurers aren’t allowed to drop drug coverage mid-year.
Finally, the agency's reforms will address a general lack of transparency in insurance plans. Insurers will be required to publish a complete list of covered drugs and their relevant cost-sharing tiers. This change will help patients better understand their plan's coverage and benefits. CMS could further improve transparency by mandating that insurers provide specific cost-sharing information for each drug.
CMS deserves praise for its newly proposed rule. It will help combat unreasonable cost-sharing arrangements, patient discrimination, and a lack of transparency. With just a few more added patient protections, the agency can ensure that quality health insurance is truly affordable for all.
Hausner is the former CEO of the American Diabetes Association.
Read More & Comment...
What do we fear more: guns being used in murder or suicide? I think the answer is obvious. And it shapes our behavior, our culture and public policy. But what is the real risk?
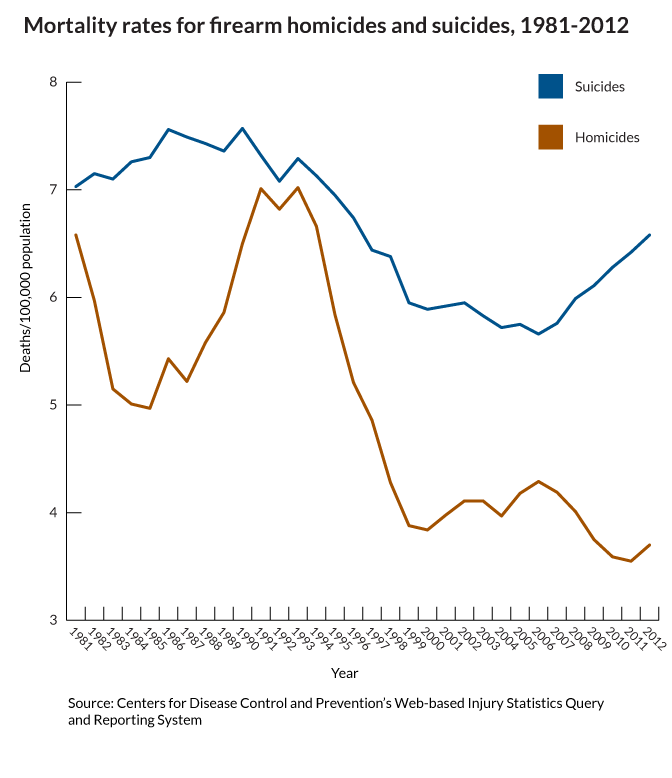
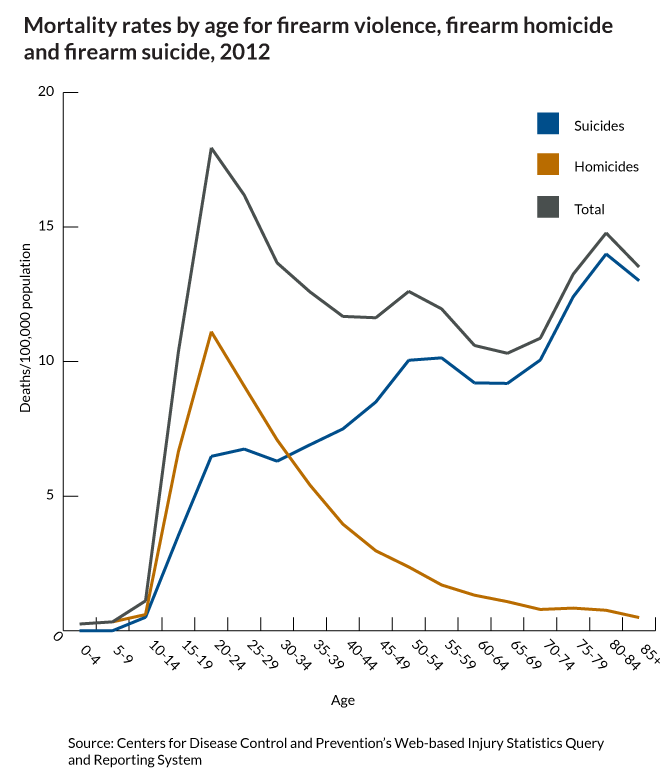
This gun violence fact is only "startling" because of how the media reinforces the fear of the unseen, outside danger...
Read More & Comment...
December 18, 2014, 02:00 pm
Closing the gap between testing and treatment
The Hill
By John Morrissey
I have just returned from San Francisco and a meeting of more than 20-thousand blood cancer experts at a global hematology conference. I was there as part of a new mission to rapidly-but-responsibly turn the data from these medical meetings into treatments.
A new alliance CLOSING-THE-GAP-NOW held a historic first conference at the University of Minnesota in mid-November. Our goal is to streamline medical oversight. I know it can be done. Under Presidents Nixon and Ford I worked to replace rules that delayed and discouraged advances in transportation.
ADVERTISEMENT
We are ready to the same for modern medicine. It is a moral imperative.
Consider. Today when a patient is prescribed a so-called new cancer treatment, it is actually the product of testing that left the laboratory as much as ten years before. That delay, the years and expense of testing, is the force driving up the costs of new therapies while denying them to patients.
The result? Tragic stories like the recent, very public case of Brittany Maynard. Diagnosed with a brain tumor and given but a few months to live, she raised the issue of the right to die. Her story really should have us thinking about what patients need to survive.
I know first hand how these lies of omission can deny hope and atrophy will to live.
I received the message that nothing can help in March 2009 when my wife contracted pancreatic cancer. Six weeks after I lost my wife, I heard that same message in October 2009 when my daughter was diagnosed with brain cancer.
The problem is, what we were told was just not the case.
In fact, there were a number of promising mainstream medical treatments in various stages of development. I found them for my family. Those innovative treatments gave us a little more time together as a family, and time is precious when it is running out.
How can we offer these same opportunities to every cancer patient?
Let’s understand that the current system is a product of the sixties. But there could be a better, faster and safer path.
Let’s harness the real power of new science, of big data, of the specificity of genome sequencing. Let’s re-imagine a review process in which each patient would get the best option and the most advanced care. Then, using computing tools with Google-like power we can capture and share all the data from their experiences. Every patient could in-effect be part of a huge virtual clinical trial while getting the best personal care.
A deluge of new information could begin to be available in ten weeks instead of ten years. Patients would not be kept waiting when they simply have no time to wait.
Thinking of tragic stories like Brittany Maynard’s, it’s long past time to appreciate the value of medical innovation and how we can use it to value each and every human life by updating the procedures and regulations that are supposed to enhance care, not interfere.
Morrissey is chair of the Scientific Advisory Board at the Richard M Schulze Family Foundation. Read More & Comment...
I’ve just returned from a visit to the UAE and Kuwait where I had the privilege of speaking with medicines regulators and pricing authority representatives from those two nations.
The topic of conversation was the value of innovation – and the urgency of rewarding it through timely approvals and appropriate pricing and reimbursement.
Our conversations ranged over many topics but focused specifically on a few key points:
* When patients have access to more effective medications, their overall health improves, even as their overall medical expenses go down. That, in turn, reduces national health-care spending and boosts the economy. Value must be measured in patient outcomes.
* Healthcare innovation saves lives, saves money, promotes economic growth, and provides hope for hundreds of millions of people (both patients and care-givers) in the United States and around the world.
* If we do not support the development of new medicines through timely licensing and fair pricing, innovation will be stopped in its tracks – and that is not an acceptable public health outcome.
* Regulators can be partners in innovation three ways: Through robust oversight, through active collaboration, and, most importantly, by being an innovation enabler.
We spent a lot of the conversation discussing the important differences between value and pricing – and that while both are important, it is value that’s the higher priority since driving patient outcomes is the higher calling (and better long-term economic investment).
After all, as Yale economist William Nordhaus has written, "The social productivity of health care spending might be many times that of other spending.”
Insha'Allah.
Read More & Comment...Emanuel noted he 'almost came to blows with the editor of the Atlantic" because he hated the title of the article. He made three important points that were not fully discussed in his article:
1. We should measure life in terms of how meaningful it is, not how long it is.
2. We should focus less on paying for end of life care and more on improving well-being in expectant mothers and adolescents.
3. We should measure meaning in terms of the capacity to create, to share and enjoy the company of others.
Ironically, the title of the article generated a lot of controversy, most likely by people who did not read his piece and used the title confirm their belief that Zeke wants to kill everyone over the age of 75. The title of the article could have been: "I Want To Live Better, Not Just Longer" but it wouldn't have created the same stir..
Read More & Comment...
"SEPTA has filed a class action suit at the U.S. District Court for the Eastern District of SEPTA logoPensylvania against Gilead Sciences, Inc. related to the sale and pricing of its Hepatitis-C drug, Sovaldi.
Sovaldi is the first drug approved by the Food and Drug Administration for certain types of Hepatitis-C infections that does not need to be injected. It can reportedly cure about 90 percent of patients with the most common form of Hepatitis-C in three to six months, and can do so with relatively minor side effects compared to earlier available treatments.
Gilead has been selling a 12-week regimen of Sovaldi in the United States for approximately $84,000, or $1,000 per pill. This is significantly more than the original price projection for Sovaldi, and in sharp contrast to the prices at which the drug is being made available in other countries, the complaint says.
Gilead recently announced its intention to make Sovaldi available in 91 developing countries at deeply discounted prices, and the drug is reportedly available in Egypt for 99 percent below the U.S. price.
While there are some orphan drugs that are similarly expensive, they are typically limited to rare conditions that affect only a very small patient population. In those instances, charging high prices may be necessary to recoup amounts invested in research and development.
In the case of Sovaldi, however, there are between 2.7 and 5.2 million people in the United States infected with Hepatitis-C, and 185 million people worldwide. The complaint alleges that, if left unchecked, Gilead’s exorbitant pricing scheme has the potential to bankrupt segments of the U.S. healthcare system."
Ok, let's go through the facts again..
The total lifetime cost of treating every HCV patient absent such innovations would be $360 billion. That does not include an estimated $400 billion loss in productivity and $3 trillion in health value lost because of premature mortality.
From this perspective it is clear that new medicines almost always reduce the cost of living longer and healthier life and increase the value of such improvements. Further, it is clear that the price of new drugs, setting aside the obvious need for faster and smarter drug development costs, reflect the high percentage of social value generated by medical innovation. Assuming the $240 billion cost goes right to innovators, more than 90% of the value of the product ($360 bill + $400 bill. + $3 trillion = $3.76 trillion) goes to society.
Read More & Comment...
Bioequivalence is going mainstream.
Ever since the FDA’s Pharmaceutical Science and Clinical Pharmacology Advisory Committee concluded in April of 2010 (in an 11-2 vote) that current bioequivalence standards are not sufficient for narrow therapeutic index (NTI) drugs, there’s been a growing focus and understanding on the fact that less variability equals better predictability.
(Narrow therapeutic index, per the FDA, means that, “small changes in blood concentration have the potential to result in serious therapeutic failures and/or serious adverse drug reactions.”)
Currently, the “sameness” of a brand product and a generic version is evaluated based on a two-treatment crossover study to prove bioequivalence, the aim being to show that the 90 percent confidence intervals of the geometric mean test/reference ratios for both maximum plasma concentration and the area under the plasma concentration-time curve fall within a range of 80 percent to 125 percent.
(“Equivalent” doesn’t mean the same thing as “identical.”)
“All men are created equivalent” just doesn’t have the same ring, does it?
This is an urgent public health issue – and it’s got momentum.
It’s too simplistic to call these “quality” problems. There’s a range from sub-standard Active Pharmaceutical Ingredient (API) and manufacturing issues, to excipient changes (excipients are the substances other than the pharmacologically active drug contained in a pill) and, most importantly, bioequivalence and bioavailability standards.
“We are losing control over what people are swallowing,” said Dr. Harry Lever, a cardiologist at the Cleveland Clinic who is trying to raise awareness of the matter among U.S. lawmakers. “Now, when a patient comes in who is not doing well, the first thing I do is look at their drugs and find out who makes it.”
Bioequivalence does not always equal therapeutic equivalence – and that’s especially true for Toprol XL. Recently two large Indian manufacturers, Wockhardt and Dr. Reddy’s Laboratories, have announced recalls over the last two months totaling more than 100,000 bottles of Toprol XL because their products were not dissolving properly — therefore probably not working as they should. The drug is a beta blocker that treats high blood pressure and heart ailments.
The issue of bioequivalence and therapeutic substitution will be at or near the top of the budget agenda in state capitols across the United States. NTI generic drugs, biosimilars and non-biologic complex drugs (NBCDs) are a medical option, but as every state in the union attempts to tighten its budget, requiring patients to use these follow-on products is an enticing, but incorrect and dangerous policy option. Placing short-term budgetary considerations before long-term patient well-being is pennywise and pound foolish, and is deleterious to both the public purse and public health. As an article in the Journal of Infection so aptly stated, “Nothing is more expensive than treatment failure.”
FDA’s recent draft guidances on bioequivalence for both generic and innovator products, as well as the move towards independent labeling for generic products are additional steps the agency has recently taken to address the issue of drug quality beyond safety and efficacy. And the implications for NTI generics, biosimilars and NBCDs is obvious.
(Something else to consider is for the FDA to report bioequivalence data in labels.)
The bioequivalence issue is clearly rising up the list of FDA priorities. Consider the agency’s action last week when it informed Mallinckrodt its methylphenidate hydrochloride tablets might not be therapeutically equivalent to Concerta, which is made by Johnson & Johnson ’s Janssen Pharmaceuticals Inc.
The FDA, in a statement, said the drug produced by Mallinckrodt may deliver the drug at a slower rate than another generic version of Concerta. As a result of its analysis, the FDA said the Mallinckrodt products are still approved and are eligible to be prescribed, but it no longer recommends them as an automatic substitution for Concerta.
But there’s a serious process issue that cannot be ignored. Mallinckrodt said the FDA informed the company that the change was based on new draft guidance for determining equivalency between the drugs that was published on November 6th – but the guidance has an open comment period that runs through January 5th, 2015. The FDA said this change was based on the application of its new Draft Guidance for determining bioequivalence of methylphenidate hydrochloride products just published on November 6, 2014. Although the Draft Guidance has an open comment period through January 5, 2015, the agency nevertheless confirmed that this change would be reflected on November 13, 2014 in the on-line Orange Book: Approved Drug Products with Therapeutic Equivalence Evaluations.
That may or may not be the proper public health decision – but it certainly doesn’t seem fair relative to thoughtful public comment. Not surprisingly, Mallinckrodt is suing the U.S. Food and Drug Administration for “unlawful” reclassification.
This is not a trivial issue. Regulatory predictability is at the foundation of the problem – and is the keystone of the solution. Minus sound and timely guidance, companies will think twice about whether or not to proceed with generic copies of medicines with a narrower therapeutic index. And that equates to less competition – which results in higher prices. This is as true for NTI medicines as it is for biosimilars. The good news is that guidances exist. The same is not true (at least not yet) for NBCDs. That should be next on the agenda – especially as it relates to A/B ratings. After all, if there are going to four categories of biosimilars, each with its significant impact on therapeutic interchangability, shouldn’t there be a commensurate finesse for similarly complex non-biologics approved under the Hatch-Waxman generic approval process?
As my friend and colleague, Dr. Scott Gottlieb has written:
The complex drugs fall in a regulatory gap. FDA has tried to retrofit the “Hatch Waxman” generic drug law and policies that govern approval of small molecule drugs to these complex drugs, with sometimes troubling results. Regardless of the decision FDA makes with Copaxone, it remains clear that Congress and FDA alike need to re-examine the regulatory process when it comes to these intricate drugs.
The problem is that FDA has refused to define these complex drugs as distinct from normal, small molecule medicines. That has forced the agency to rely on less information in approving these complex copies than it probably would like. The agency’s desire to try and squeeze these complex drugs through its existing generic law approval pathway may have as much to do with political expediency as with good science. FDA is probably well aware that getting Congress to define a distinct category for these medicines, and give FDA proper tools, could be a heavy political lift. So FDA is doing what it often does: trying to massage its existing authorities and regulatory practices to fit novel challenges. But at what cost?
The good news is that the FDA recognizes the need for more specific, regular, and predictable standards. That’s precisely why the agency has asked industry to identify drug products that require explicit bioequivalence testing guidances.
Robert Lionberger, head of the FDA’s Office of Research and Standards, said the agency’s objective is to develop a product-specific BE guidance before a manufacturer files an ANDA for the drug in question.
That bad news, per a report in Drug Industry Daily, is that the agency has more than 1,200 product-specific guidances posted to aid ANDAs, leaving hundreds of potential generic targets without an FDA-sanctioned roadmap for development. Their absence has complicated the process of submitting successful ANDAs.
Is the NBCD pathway, for example, something that should be considered as part of the pending 21st Century Cures legislation?
We are increasingly living in an n-of-1 world. Small is the new Big. We must think differently about bioequivalence on the front end and pharmacovigilance on the back-end. While we must continue to capture adverse event data, we must also strive to capture Substandard Pharmaceutical Events (SPEs). SPEs occur when a product does not perform as expected—perhaps because of API or excipient issues – or too broad of a bioequivalence range and issues relating to therapeutic interchangeability.
When it comes to 21st-century pharmacovigilance, we have to both broaden and narrow our views about bioequivalence to the patient level. Outcomes is so much more than a value-based reimbursement issue.
Read More & Comment...There is a growing awareness that science of attacking cancer is reaching a critical mass and is producing a truly transformational approach to treatment: one that re-engineers our natural immune responses to do the job of not just killing cancer cells but teaches the remaining ones to start become healthy.
There is a growing awareness that the way insurers have shifted cost to patients is discriminatory and that the claim that it’s a reaction to high prices is bogus.
There is a growing recognition that the value new medicines yield must be measured in terms of every month of every life gained. More people are alive today because each era of treatments keeps people alive longer for the next generation of therapies that improve and lengthen life even more.
And there is a growing recognition that the pace of progress is too damn slow, too dependent on outdate scientific methods that require people dying of disease to be randomized to different treatments or different combinations of treatments in ways that have little to do with their unique and increasingly accessible biological differences.
There is an increasing emphasis on improving the quality of life and the fact being treated for cancer does not mean being treated like a victim.
And finally, people are increasingly aware that by sharing information more quickly about the real world experience with medicines is the most scientific and efficient way to cure cancer.
I am honored to be part of this courageous community of advocates, patients, scientists and biopharmaceutical companies that express their awareness with growing discontent with the status quo. As Thomas Edison said: “Discontent is the first necessity of progress.”
Read More & Comment...
At the same time that regulators in France, Germany, Belgium and Luxembourg are suspending the marketing approval of 25 generic drugs due to concerns over the quality of data from clinical trials conducted by India's GVK Biosciences, another Indian firm, Zydus Cadila, has launched a biosimilar of Adalimumab (an anti-TNF-α monoclonal antibody, which is approved in many countries for the treatment of inflammatory diseases, including rheumatoid arthritis, plaque psoriasis, polyarticular juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn's disease and ulcerative colitis.)
Bad timing and, perhaps, bad science.
The quality of Indian pharmaceuticals has come under fire this year, with regulators in Europe and the United States citing problems ranging from data manipulation to sanitation and banning the import of certain products from several firms. And, it should be noted, these violations are for generic drugs, not biosimilars.
French and German regulators are investigating drug approvals based on clinical trials meant to show that these generic drugs were equivalent to the original branded versions conducted by GVK Bio between 2008 and 2014.
A Zydus Cadila spokesman commented that, “This therapy will offer a new lease of life to millions in India who have not had access to this therapy so far.” But the reality is that their product is more likely to be aggressively marketed for export.
Most importantly is the question of quality – or lack thereof. As company’s ranging from biotech innovator Amgen to multinational generics manufacturer Sandoz struggle to fulfill the clinical requirements for an Adalimumab biosimilar, two obvious questions arise -- (1) What does Zydus Cadila know that large multinational manufacturers don’t; and (2) What do Indian regulators know that their counterparts in the US and the EU are struggling to understand?
To not aggressively ask these questions is to bury one’s head in the sand.
And here’s an even tougher question -- In order to boost pharmaceutical exports, are Indian regulators willing to license substandard products?
According to a May 2012 article in The Lancet,
“To say that India's drug regulatory authority, the Central Drugs Standard Control Organisation (CDSCO)-whose remit includes new drug approval, licensing of manufacturing facilities, and regulation of drug trials-is not fit for purpose seems a gross understatement.”
“If I have to follow U.S. standards in inspecting facilities supplying to the Indian market,” G. N. Singh, India’s top drug regulator, said in an interview with an Indian newspaper, “we will have to shut almost all of those.”
Does that mean US standards are too high or that Indian ones are too low? Well, where you stand depends on where you sit. And if you’re sober and sitting up straight, the answer is obvious.
According to Dilip Shah, Secretary General of the Indian Pharmaceutical Alliance, some Indian manufacturers “doctor their data.” The solution? “Regulators should have regular liaison with manufacturers in India and China to explain to them how GMP works and that they don’t have to cheat.”
Feel better now?
Read More & Comment...The bad news is that Missouri is the only state that does not have a prescription drug monitoring program.
According to the New York Times, Missouri is the only state in America that has declined to keep a prescription drug database — the primary tool the other 49 states use to identify people who acquire excess prescriptions for addictive painkillers and tranquilizers, as well as the physicians who overprescribe them … Not having the database has not only hampered Missouri’s ability to combat prescription drug abuse, but also attracted people from neighboring states looking to stockpile pills and bring them home to take themselves or sell to others, according to law enforcement officials, legislators and data compiled by a prescription drug processing firm.
“Welcome to Missouri — America’s Drugstore,” said Dr. Douglas Char, an emergency room physician in St. Louis. “We aren’t just allowing abuse, we’ve created a business model for dealers.”
The good news is that Republican State Representative Holly Rehder is drafting a bill to change that.
The prescription drug-monitoring program (PDMP) is a database that collects information to let doctors and pharmacists lets doctors and pharmacists know a patient's prescription history. The program is used to reduce the amount of medications that are sold on the street and to reduce the risk of doctor shopping and abusing painkillers for nonmedical reasons.
One of the reasons the Show Me State has remained the Don't Show me State are unfounded concerns over privacy and Big Brotherism. Rehder’s proposed program would allow for only doctors and pharmacists to obtain the information and police officers would have to obtain a warrant to access the information.
Bob Twillman from the American Academy of Pain Management said the program helps identify not only if people are abusing drugs, but identifying their addiction so they can get professional help.
The bill previously passed in the House but has run into some roadblocks in the Senate, including St. Joseph Senator Rob Schaaf, who said the database would violate the privacy of citizens. After successfully sinking a 2012 version of the bill, Mr. Schaaf said of drug abusers, “If they overdose and kill themselves, it just removes them from the gene pool.”
Really?
Rehder said she is willing to work with Schaaf and others in order to get the bill passed.
Holly – don’t go lightly.
Read More & Comment...ACA should allow prescription drugs
President Barack Obama is fully vested in the claim that his Affordable Care Act is a success.
During a recent news conference, he pointed to the supposedly salutary effect the ACA has had on mitigating rising health care costs. "Health care inflation has gone down every single year since the law passed, so that we now have the lowest increase in health care costs in 50 years," Obama said.
Obama is right that health care inflation has slowed. The Centers for Medicare and Medicaid Services have reported exactly that for the past several years. But his praise is misplaced. Thanks is not due to the ACA but rather to increased access to prescription drugs, which help folks manage illnesses and stay out of the hospital. The ACA actually threatens to increase health care spending by keeping these vital prescription drugs out of reach for many Americans.
If policymakers are serious about slowing runaway health care costs, they need to ensure that sick people have good access to prescription medicines that will make them better.
Consider the example of Medicare Part D. In 2006, Medicare Part D expanded access and reduced the costs of prescription drugs for millions of Americans. The following year, prescriptions filled by Medicare beneficiaries jumped by 14 percent, and, as a result, the annual growth in spending per beneficiary dropped by almost half.
A new National Bureau of Economic Research study credited Part D coverage with an 8 percent drop in hospital admissions for seniors and around $1.5 billion in reduced hospital spending.
The Congressional Budget Office reached a similar conclusion in 2012, finding that spending on medical services dropped by 0.2 percent for every 1 percent increase in prescriptions filled.
In fact, failure to regularly take prescribed medications puts considerable strain on the health care system. A study from the Annals of Internal Medicine estimates that non-adherence already costs the health care system $100 billion to $289 billion a year. That includes avoidable hospitalizations, nursing home admissions and premature deaths.
Given the power of prescription drugs, the Obama administration should be pushing to increase their accessibility. Instead, the administration is actively working against them.
Since Medicare Part D began, the government has required insurers to cover drugs in six treatment areas. The Obama administration, however, proposed early this year to limit coverage of immunosuppressants, antidepressants and antipsychotic medicines for diseases like schizophrenia. The proposal caused an outcry from patients, health care providers and members of both political parties, and the administration scrapped the plan.
But now the Obama administration is doing damage elsewhere — by limiting prescription drugs through the health care exchanges created by the ACA.
Plans available through the exchanges often charge patients significant out-of-pocket fees to access needed prescription drugs. For moderate to low-income patients, these high costs may force them to miss prescriptions.
Insurance companies decide how to share costs with patients by using a tiered system. A patient who needs a drug in the lowest tier might have to pay only a $15 copay. But for those drugs in the highest tier, patients can pay as much as 40 percent of the actual cost of the drug, which can run up to tens of thousands of dollars.
Too many key medications on too many plans are now listed in the highest tier. A recent study from Avalere Health found that more than a fifth of all "Silver" Plans — the most popular coverage — require patients to pay 40 percent of the costs associated with seven classes of drugs. And 60 percent of Silver Plans placed all drugs for multiple sclerosis and rheumatoid arthritis on the highest cost-sharing level.
Just as expanded access to drugs reduces overall health care spending, restricting access drives up costs. Patients who can't readily afford to spend thousands of dollars on, say, their AIDS medication, will probably end up skipping doses. This non-adherence will increase hospitalization rates and overall health care spending.
If the Obama administration wants to put a brake on health care spending, it must preserve robust access to prescription drugs. The White House's attempts to limit prescription drug access in Medicare Part D are incredibly shortsighted. Instead, the administration should be doing all it can to ensure that more Americans have access to these vital drugs.
Peter J. Pitts, a former Food and Drug Administration associate commissioner, is president of the Center for Medicine in the Public Interest.
Read More & Comment...Here's a snapshot of what the impact HIV would have today on hospitalization, hospital costs and hospital deaths if we had listened to the naysayers. Data for chart below is from Agency for Healthcare Research and Quality (AHRQ), based on data collected by individual States and provided to AHRQ by the States for the Clinical Classification Code for HIV
 Read More & Comment...
Read More & Comment...
Eleven of the 12 cancer drugs the Food and Drug Administration approved for fighting cancer in 2012 were priced at more than $100,000 per year, double the average annual household income, according to a report by the Journal of National Cancer Institute.
Those claims are inaccurate at best and lies at worst.
First, as a Milliman study showss, the average cost of chemotherapy is about $25000. A far cry from the $120K Kantarjian and Bacn assert.
And while most of the new targeted cancer drugs are expensive, the average price -- including a standard discount and not counting any patient assistance -- is $5000 a month.
The fact is, cancer drugs, as a percentage of what people spend, of cancer care and total health care spending is small.

An HHS study shows that prescription drugs, on average, cost people with cancer about $800 a year.
Kantarjian and others claim cancer drug prices cause bankruptcy. Also misleading. Cancer patients filing for bankruptcy comprise 2.2 percent of total filings. That's about 36,000 people a year. The vast majority are low income and are unemployed. That is a financially crushing combination for many more cancer patients beyond those who file for bankruptcy. But drug prices are not to blame.
It is true that many more people are paying thousdands for cancer drugs but as my next blog reveals the Kantarjians and Bachs of the world deliberately ignore the role insurance comapne play in turning life saving drugs into a finanially sickening challenges. More on that in my next post.
Read More & Comment...
This Sunday, the Boston Globe asked, “Has health care improved with reclassification of hydrocodone?”
Here’s the response of the dynamic Cindy Steinberg, policy chair for the Massachusetts Pain Initiative and national director of policy and advocacy for the US Pain Foundation:
No.
Severely restricting access to the most commonly used and highly effective pain medication is unjustly punishing the millions of law-abiding citizens who struggle to live with pain every day while doing little to solve the problem of abuse and addiction.
Hydrocodone combination products are effective for both acute and chronic pain and are useful and appropriate for a wide range of painful conditions and diseases. They have relatively few side effects and have been shown to have lower abuse potential than single entity opioids. In 2011, approximately 47 million Americans used hydrocodone analgesics for pain relief.
The number of Americans now living with pain is staggering. The Institute of Medicine has documented that there are more than 100 million living with chronic pain in this country. Ten million Americans live with chronic pain so severe that it has disabled them, and this number is expected to increase as the population ages.
When prescribed to individuals with pain severe enough to require these medications, the incidence of abuse and addiction is extremely low. For many with chronic pain, these medications mean the difference between a life worth living or not.
The new rules took effect Oct. 6. They require patients to obtain an original hard copy prescription for every 30 days of their medication — necessitating many more doctor visits — and then hand-delivering the script to their pharmacy. Doctors can no longer call in, fax, or electronically submit these prescriptions. Refills are prohibited.
According to Rebecca Fortelka, who suffers from cerebral palsy and several other chronic pain conditions writing in the Nov. 18 National Pain Report, hydrocodone combinations are one of the only pain medications she can tolerate. Soon after the new rules went into effect, she contacted her doctor’s office as she was running out of her medicine and needed a renewal. “My pain was spiraling out of control as I was rationing meds to prevent being out of them,” she wrote. It took her doctor’s office four weeks to get the hard copy prescription ready, leaving her to suffer.
Cutting off the supply of pain medication will not solve abuse and addiction. Those who are predisposed to the disease of addiction will turn to other medications or illicit substances, leaving those with the disease of chronic pain with fewer options. People with addiction disorders need better treatment for the disease of addiction, coordinated follow-up, and continuing support.
Read More & Comment...
Recent critics of cancer drug prices claim that the new higher priced medications drive up total health care spending.
In fact, the opposite is the case. The evidence is overwhelming. In fact, the addition of new medicines under Medicare's drug plan (Part D) has led to an absoute reduction in what's been spent on Medicare overall, $380 billion less to be exact. A recent Health Affairs blog notes: "Much attention in particular has focused on the remarkable slowdown in Medicare spending over the past few years, and rightfully so: Spending per beneficiary actually shrank (!) by one percent this year (or grew only one percent if one removes the effects of temporary policy changes).
Yet the disproportionate role played by prescription drug spending (or Part D) has seemingly escaped notice. Despite constituting barely more than 10 percent of Medicare spending, our analysis shows that Part D has accounted for over 60 percent of the slowdown in Medicare benefits since 2011 (beyond the sequestration contained in the 2011 Budget Control Act)."

Critics will respond that the decline in Medicare spending was a function of the recession. Not true. "The recession and its aftermath appear to have had little effect on Medicare Part A and Part B spending; senior’s incomes are less influenced by economic downturns and the large majority of beneficiaries have supplemental coverage that shields them from most Part A and Part B cost-sharing. The primary analysis on the topic from CBO, in fact, found that only one-eighth of the slowdown in Parts A and B from 2007-2010 could be explained by factors related to the economy – and not through the usual channel of utilization impacts that take place in the private market."
Finally, some argue that the decline in spending was due to increased use of generic drugs on the part of Medicare consumers. In fact, most of the increase in Part D spending is the result of the use of new medicines for cancer, MS, diabetes and other drugs for the most expensive conditions Medicare patients deal with. This is in line with studies conducted by Frank Lichtenberg and others showing that each dollar spent on new medicines reduces what would be spent on hospitals, doctors, etc. by $6.
Looking at the price of a new medicine or what we spend on biopharmaceuticals in isolation is misleading. Use of innovative medicines should be looked at in terms of the impact it has on other medical spending and the lives of patients.
Social Networks
Please Follow the Drugwonks Blog on Facebook, Twitter, LinkedIn, YouTube & RSS
Add This Blog to my Technorati Favorites