DrugWonks on Twitter
Tweets by @PeterPittsDrugWonks on Facebook
CMPI Videos

Video Montage of Third Annual Odyssey Awards Gala Featuring Governor Mitch Daniels, Montel Williams, Dr. Paul Offit and CMPI president Peter Pitts

Indiana Governor Mitch Daniels

Montel Williams, Emmy Award-Winning Talk Show Host

Paul Offit, M.D., Chief of the Division of Infectious Diseases and the Director of the Vaccine Education Center at the Children’s Hospital of Philadelphia, for Leadership in Transformational Medicine

CMPI president Peter J. Pitts

CMPI Web Video: "Science or Celebrity"
Tabloid Medicine

Check Out CMPI's Book
A Transatlantic Malaise
Edited By: Peter J. Pitts
Download the E-Book Version Here
CMPI Events

Donate

CMPI Reports

Blog Roll
AHRP
Better Health
BigGovHealth
Biotech Blog
BrandweekNRX
CA Medicine man
Cafe Pharma
Campaign for Modern Medicines
Carlat Psychiatry Blog
Clinical Psychology and Psychiatry: A Closer Look
Conservative's Forum
Club For Growth
CNEhealth.org
Diabetes Mine
Disruptive Women
Doctors For Patient Care
Dr. Gov
Drug Channels
DTC Perspectives
eDrugSearch
Envisioning 2.0
EyeOnFDA
FDA Law Blog
Fierce Pharma
fightingdiseases.org
Fresh Air Fund
Furious Seasons
Gooznews
Gel Health News
Hands Off My Health
Health Business Blog
Health Care BS
Health Care for All
Healthy Skepticism
Hooked: Ethics, Medicine, and Pharma
Hugh Hewitt
IgniteBlog
In the Pipeline
In Vivo
Instapundit
Internet Drug News
Jaz'd Healthcare
Jaz'd Pharmaceutical Industry
Jim Edwards' NRx
Kaus Files
KevinMD
Laffer Health Care Report
Little Green Footballs
Med Buzz
Media Research Center
Medrants
More than Medicine
National Review
Neuroethics & Law
Newsbusters
Nurses For Reform
Nurses For Reform Blog
Opinion Journal
Orange Book
PAL
Peter Rost
Pharm Aid
Pharma Blog Review
Pharma Blogsphere
Pharma Marketing Blog
Pharmablogger
Pharmacology Corner
Pharmagossip
Pharmamotion
Pharmalot
Pharmaceutical Business Review
Piper Report
Polipundit
Powerline
Prescription for a Cure
Public Plan Facts
Quackwatch
Real Clear Politics
Remedyhealthcare
Shark Report
Shearlings Got Plowed
StateHouseCall.org
Taking Back America
Terra Sigillata
The Cycle
The Catalyst
The Lonely Conservative
TortsProf
Town Hall
Washington Monthly
World of DTC Marketing
WSJ Health Blog
DrugWonks Blog
According to Peggy Hamburg ...
The difference between science and science fiction is a line that seems ever harder to distinguish, thanks in part to a host of astonishing advances in medical science that are helping to create a new age of promise and possibility for patients.
Today cancer drugs are increasingly twinned with a diagnostic device that can determine whether a patient will respond to the drug based on their tumor’s genetic characteristics; medical imaging can be used to identify the best implantable device to treat a specific patient with clogged coronary arteries; and progress in regenerative medicine and stem cell therapy using a patient’s own cells could lead to the replacement or regeneration of their missing or damaged tissues. Given these trends, the future of medicine is rapidly approaching the promising level of care and cure once imagined by Hollywood in futuristic dramas like Star Trek.
But these examples are not science fiction. They are very real achievements that demonstrate the era of “personalized medicine” where advances in the science of drug development, the study of genes and their functions, the availability of increasingly powerful computers and other technologies, combined with our greater understanding of the complexity of disease, makes it possible to tailor treatments to the needs of an individual patient. We now know that patients with similar symptoms may have different diseases with different causes. Individual patients who may appear to have the same disease may respond differently (or not at all) to treatments of that disease.
FDA has been playing a critical role in the growth of this new era for a number of years. Even before I became FDA Commissioner the agency was creating the organizational infrastructure and putting in place the regulatory processes and policies needed to meet the challenges of regulating these complex products and coordinating their review and oversight. It has been my pleasure to serve at FDA during this next exciting period and to help ensure that the agency continues to prioritize this evolution by anticipating, responding to, and encouraging scientific advancements.
I am very pleased to be able to present a new report by FDA as part of our ongoing efforts in this field. Paving the Way for Personalized Medicine: FDA’s Role in a New Era of Medical Product Development describes many of the exciting developments and looming advances in personalized medicine, lays out the historical progress in this field, and examines FDA’s regulatory role: from ensuring the availability of safe and effective diagnostic devices, to addressing the challenges of aligning a drug with a diagnostic device, to post-market surveillance.
Outside collaboration and information sharing is essential for this field to flourish. On Tuesday, the American Association for Cancer Research and AdvaMedDX held a fruitful daylong conversation on personalized medicine to treat cancer. I was one of the speakers, participating in a conversation with Dr. Francis Collins, the head of the National Institutes of Health. Our discussion focused in part on current status of drug and diagnostic co-development and the challenges and potential of whole genome sequencing, where data can be collected on a patient’s entire genetic makeup at a reasonable cost in a reasonable amount of time.
FDA is committed to fostering these cooperative efforts, as it will require the full force of government, private industry, academia and other concerned stakeholders to maximize our efforts and fully realize the promise of personalized medicine. Our new report outlines that commitment, and helps chart the way forward so that more people can live long and prosper.
Read More & Comment...Via the New Republic:
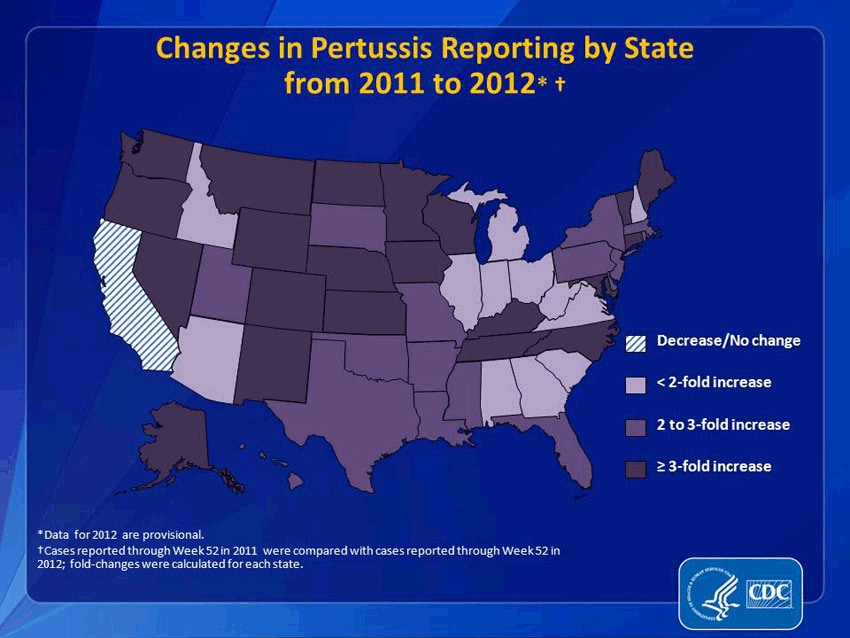
It would be an understatement to say that pertussis and other formerly conquered childhood diseases like measles and mumps are making a resurgence. Pertussis, specifically, has come roaring back. From 2011 to 2012, reported pertussis incidences rose more than threefold in 21 states. (And that’s just reported cases. Since we’re not primed to be on the look-out for it, many people may simply not realize they have it.) In 2012, the CDC said that the number of pertussis cases was higher than at any point in 50 years. That year, Washington state declared an epidemic; this year, Texas did, too. Washington, D.C. has also seen a dramatic increase. This fall, Cincinnati reported a 283 percent increase in pertussis. It’s even gotten to the point that pertussis has become a minor celebrity cause: NASCAR hero Jeff Gordon and Sarah Michelle Gellar are now encouraging people to get vaccinated.
{kind=link}
How responsible are these non-vaccinating parents for my pertussis? Very. A study recently published in the journal Pediatrics indicated that outbreaks of these antediluvian diseases clustered where parents filed non-medical exemptions—that is, where parents decided not to vaccinate their kids because of their personal beliefs. The study found that areas with high concentrations of conscientious objectors were 2.5 times more likely to have an outbreak of pertussis. (To clarify: I was vaccinated against pertussis as a child, but the vaccine wears off by adulthood, which, until recently, was rarely a problem because the disease wasn't running rampant because of people not vaccinating their kids.)
So thanks a lot, anti-vaccine parents. You took an ethical stand against big pharma and the autism your baby was not going to get anyway, and, by doing so, killed some babies and gave me, an otherwise healthy 31-year-old woman, the whooping cough in the year 2013. I understand your wanting to raise your own children as you see fit, science be damned, but you're selfishly jeopardizing more than your own children. Carry your baby around in a sling, feed her organic banana mash while you drink your ethical coffee, fine, but what gives you denialists the right to put my health at risk—to cause me to catch a debilitating, humiliating, and frightening cough that, two months after I finished my last course of antibiotics (how’s that for supporting big pharma?), still makes me convulse several times a day like some kind of tragic nineteenth-century heroine?
If you have an answer, I’ll be here, whooping, while I wait.
Read the full article here.
Read More & Comment...
According to a new letter to the British Medical Journal, “Doctors may not report adverse events or speak up when they witness poor care because of fear of punitive action or lack of confidence that reporting will change anything.”
Whose responsibility is it to build confidence in both the pharmacovigilance process as well as the urgent importance of the proposition? I believe it’s the job of the regulatory body that oversees the both the procedures and the actions that derive from post-marketing reports. That means (in the US), the FDA.
Would it be nice if the FDA could get additional dollars to ramp up pharmacovigilance beyond MedWatch and Sentinel? Sure, but let’s get real – that ain’t gonna happen. What the FDA does have at its disposal is the bully pulpit. It’s time for Janet, Peggy, and Gerald to get up on their soapboxes and start preaching the urgency and importance of pharmacovigilance.
Such renewed efforts are called for since the focus is now increasingly on patient outcomes. If we can’t measure it, it doesn’t count – or counts for less. Perhaps it’s time for the FDA to create an advisory committee of pharmacovigilance issues – and hold meetings to focus on process improvement, greater stakeholder involvement (not just with physicians, but with pharmacists, hospitals and patients), and better ways to share post-marketing data in a transparent and timely fashion – and not just adverse events and label updates but also Substandard Pharmaceutical Events (SPEs), when patient’s don’t respond as they should when their therapy has been impacted by therapeutic switching or interchangeability/bioequivalence issues.
Can you say, “biosimilars?” It's an adverse event horizon.
Read More & Comment...Sebelius & Co. have released enrollment numbers. Some key findings:
* 106,185 people have selected an Exchange plan (although not all have paid the premium). This includes 26,794 people in the federal exchange states.
* 396,261 people whose applications were processed by the Exchanges have been deemed eligible for Medicaid. A little more than half of those people live in states with state-based exchanges.
* Surprisingly, of those deemed eligible for Marketplace plan enrollment, only 30% qualified for a premium tax credit. CBO had expected about 80% of Exchange enrollees would qualify for a premium tax credit.
* About a million people have been deemed eligible for Exchange coverage but have not yet picked a plan. It’s not clear if these people will pick a plan later on or decided not to enroll – especially if their “cancelled” plans are reinstated.
* The HHS release includes a long appendix comparing Exchange enrollment to Part D, Massachusetts Commonwealth Care, CHIP and FEHBP. While Part D had about 10% enrollment in the first month, Massachusetts had just 3% enrollment in the first month (and the Bay State kept enrollment open for the entire first year so there was less pressure to enroll early).
* If exchange enrollment mirrors the same enrollment pattern as Part D, about 700,000 people would have to enroll by the end of November in order to meet the target of 7,000,000 people enrolled in Exchanges in 2014.
And speaking of exchanges, Gallup has found that just 18% of uninsured adults had attempted to visit an Exchange website.
Technical notes:
* This report only captures enrollment through Nov 2.
* Data includes applications submitted through methods other than the website (i.e. paper applications).
* There are state-by-state tables included in the release (see page 9).
The complete HHS release can be found here.
Read More & Comment...The term “context of use” refers to a comprehensive description that fully and clearly delineates the limits of FDA’s qualification decision in terms of the manner and purpose of use for the DDT(s). The context of use statement should describe all criteria under which the DDT is qualified for use. The qualified context of use defines the boundaries within which the available data adequately justify use of the DDT(s). As data from additional studies are obtained over time, submitters of DDTs may continue working within the DDT Qualification Programs to submit additional data and expand the qualified context of use.
http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugDevelopmentToolsQualificationProgram/
What is Qualification?
Qualification is a conclusion that within the stated context of use, the DDT can be relied upon to have a specific interpretation and application in drug development and regulatory review. Once qualified, DDTs will be publicly available to be used in any drug development program for the qualified context of use. The qualified DDT can be included in IND or NDA/BLA submissions without the need for CDER to reconsider and reconfirm the suitability of the DDT as long as:
* There are no serious study flaws
* There are no attempts to apply the DDT outside the qualified context of use
* There are no new and conflicting scientific facts not known at the time the qualification was determined
Qualification may contribute to acceptance and application of DDTs across multiple drug development programs. Having qualified DDTs that can be utilized by many sponsors will aid in optimizing drug development and evaluation.
These Qualification Programs promote a collaborative setting in which there are advantages for multiple interested parties to work together in a consortium to develop DDTs for qualification. Resources and knowledge-sharing outside a specific drug development program can accelerate DDT development and facilitate availability of DDTs at critical milestones for future drug development programs. Early and continued interactions with FDA during the DDT development process are critical to the success of these programs.
Regulators love ambiguity because it gives them power. But flexibility is important. All this to say, it’s a tough balance.
And, once again, context matters.
Read More & Comment...
From the pages of The Washington Examiner ...
Here comes the next big healthcare disaster, courtesy of HHS
And you thought the rollout of Obamacare was a catastrophe?
Here’s an immediate and critical pending national healthcare disaster — a new flu pandemic with nobody home at the Department of Health and Human Services.
This past April, a new strain of H7N9 avian influenza emerged in China and quickly spread throughout the region.
Though the H7N9 virus was only known to transfer directly from an avian source, numerous people were exposed to the disease’s devastating illnesses.
By the time the virus had momentarily vanished in August, a staggering 44 of the 134 infected had died, a 33 percent mortality rate.
At first, HHS took the necessary steps to address the new pandemic and prepare for the worst. Health and Human Services Secretary Kathleen Sebelius determined that the virus was a threat to U.S. national security and to Americans living abroad.
Subsidiary offices of HHS, the Biomedical Advanced Research and Development Authority and the Centers for Disease and Control, recommended producing a seed vaccine, conducting clinical trials and, if the threat of pandemic remained, contracting manufactures to produce vaccine stockpiles.
Once the virus was identified as a legitimate threat, HHS moved quickly to contract with vaccine manufacturers to produce a vaccine and prepare for mass production if the virus was to resurface, as influenza viruses typically do.
It seemed as though the government was taking on this threat appropriately — until Sebelius’ slogging bureaucracy, those wonderful people who brought you Obamacare, took over.
In September, BARDA contracted three vaccine manufacturers for $110 million to put in order a vaccine and prepare mass vaccine stockpiles once HHS gave them the go-ahead.
Reportedly, HHS was supposed to provide the manufacturers with further instructions by mid-October.
Yet today, HHS still sits on the very production orders that are necessary to stockpile the H7N9 vaccine to protect against a pandemic.
HHS’s timing could not be worse, with flu season already upon us. As of this week, there have been four newly reported cases of the H7N9 virus in China, leading a spokesman for the World Health Organization to conclude succinctly: "Winter is starting."
While experts have stopped short of guaranteeing a spike in H7N9 cases, the threat of a pandemic is anything but far-fetched.
If the second phase of this virus evolves from an avian-transferred virus to a human-to-human transfer, we will see devastating results.
Perhaps staff at HHS should heed the words of their colleague, Dr. Tom Frieden, director of the CDC.
In September, Frieden warned that “nothing has the potential to kill more people than influenza. A strain such as H7N9 could create a pandemic. When something with that much potential to cause widespread harm emerges, we must identify it in hours or days, not weeks or months.”
It will take, at minimum, 60 days to produce the first wave of vaccine stockpiles once an order is given to vaccine manufacturers.
And that is just the first wave, a quantity that would be wholly insufficient for the American public at large. Protecting the entire U.S. population would take many months, time that, frankly, we may not have.
Unfortunately, now HHS has again demonstrated gross incompetence in its obligation to best promote the public health. Its eye is not on the prize of pandemic preparedness.
If the federal bureaucracy continues to stall on producing the H7N9 vaccine, HHS could face a catastrophe that would make the healthcare.gov storm seem like a light spring shower.
Peter Pitts, a former FDA associate commissioner, is president of the Center for Medicine in the Public Interest.
Read More & Comment...From the pages of Law360.com …
Iclusig Deaths Won't Stall FDA's Speedier Drug Approvals
By Jeff Overley
The jarring death toll linked to leukemia drug Iclusig will force the U.S. Food and Drug Administration to revisit the science that won the product accelerated approval, but a growing willingness of seriously ill patients to accept risk means the tragic episode is unlikely to delay other fast-tracked medicines, experts say.
Sales of the chemotherapy drug, which earned FDA clearance in December, were halted last week after manufacturer Ariad Pharmaceuticals Inc. reported that at least 14 fatalities had occurred among 530 patients in two clinical trials. In addition, scores of other people suffered major side effects resulting from blood clots, including tissue death that led to amputations.
The dramatic circumstances add an important chapter to a long-running debate about FDA’s various “priority review” programs, which involve quicker analysis by regulators and may apply a relaxed safety standard.
While FDA is wary of risky medications, it’s increasingly under pressure from patients who say potentially dangerous treatments for deadly diseases are better than no treatments at all. That’s a trend with roots in the darkest days of the HIV/AIDS crisis, and it has only picked up steam in recent years.
“If someone has no options, it makes sense to try anything,” said Areta L. Kupchyk, a partner at Nixon Peabody LLP and former associate chief counsel for drugs and biologics at FDA.
Many drugs, even those approved on traditional pathways, are found to have more serious risks post-approval, but the Iclusig saga is particularly notable, as FDA rarely urges manufacturers to pull products from the market.
The development also comes at a time of rising scrutiny of whether the FDA has found the sweet spot in addressing unmet medical needs while continuing to protect patients from toxic medicines.
“It’s a very precarious balance,” said Peter J. Pitts, president and co-founder of the nonprofit Center for Medicine in the Public Interest.
For example, scholarly papers have explored whether standards are too loose. One article, published last year in the Journal of the American Medical Association, called particular attention to three products — AstraZeneca PLC’s cancer drug Caprelsa, Novartis AG’smultiple sclerosis medicine Gilenya, and Boehringer Ingelheim GmbH’s blood thinner Pradaxa — and asked whether their risk-benefit profiles made priority review inappropriate.
The safety record of the drugs raises the question “of whether it was good policy to approve three innovative new drugs with significant safety questions unanswered and with optimal doses not determined," according to the researchers Thomas Moore of the Institute for Safe Medication Practices and Dr. Curt Furberg, then of the Wake Forest University School of Medicine.
In a second article published just days before the freeze on Iclusig sales, the same authors found that drugs receiving expedited approval in 2008 typically used data from trials with less than one-fifth the usual number of patients and that progress was slow on completing mandatory post-approval studies.
In some recent years, nearly half the new drugs cleared by the FDA have used expedited pathways, raising the stakes should those routes be called into question by a review of Iculsig's approval.
“The question one has to ask is: Were there ... safety signals before this?” Kupchyk said. “That might be something that FDA and others wants to look at more closely.”
But while the events surrounding Iclusig are serious, experts say they don’t necessarily reflect poorly on priority review. FDA knew that blood clots were a risk at the time of approval and required a black box warning to that effect. It also ordered post-approval analysis that discovered greater-than-expected dangers less than one year after clearance, suggesting things worked largely as designed.
“What happened here seems to be consistent with the program, and I don’t see that FDA is going to pull back necessarily unless there is some evidence that they missed,” Kupchyk said.
FDA’s reaction with respect to its overall approach to expedited approvals is hard to predict because the amount of acceptable risk can vary greatly depending on which condition is being treated and whether there are any other effective drugs available.
“Safe is a relative concept. You wouldn’t approve a drug for allergies that is fatal for 30 percent of patients,” Pitts said.
Also, the prerogatives of distinct patient communities are influencing FDA more and more. Last year’s user-fee law directed the agency to refine its risk-benefit assessments, and a big part of that is so-called patient-focused drug development, which entails staging public meetings to gauge willingness to gamble on unproven drugs. Gatherings so far have covered chronic fatigue syndrome, HIV, lung cancer and narcolepsy, and more will take place involving fibromyalgia, sickle cell anemia and more than a dozen other conditions.
On Wednesday, an FDA spokeswoman defended the current system and did not directly answer a question about how the agency would decide whether any mistakes were made in approving Iclusig, which remains available in extremely narrow circumstances.
“We have a robust program for post-marketing surveillance and ensuring the benefits of a marketed drug outweigh its risks," FDA spokeswoman Stephanie Yao said. "If that profile changes in any way, we review and take appropriate action."
Regardless of whether anything should have been done differently with Iclusig, when dealing with the deadliest diseases and strong lobbying from people affected by them, experts say it may be inevitable that things occasionally go awry.
“When it comes to accelerated approval, it’s a high-risk, high-reward situation,” Pitts said. “The FDA’s not going to get it right all the time.”
Read More & Comment...From the pages of the New York Times …
Label Updates May Be Allowed for Generics
By KATIE THOMAS
The Food and Drug Administration proposed a rule on Friday that would permit generic drug makers to update their labels if they received information about potential safety problems. The move puts the companies on equal footing with brand-name manufacturers, but it also opens the door to lawsuits against them for the first time since the Supreme Court barred such cases two years ago.
Until now, only brand-name drug makers had the ability to independently update their labels if important new information surfaced about one of their drugs. If the F.D.A. agreed that the new information warranted a change in the label, then generic companies were required to update their product information so that it would be identical to the branded drug’s labeling.
Dr. Janet Woodcock, the F.D.A.’s head of drug evaluation and research, said the proposed change would create better parity between brand-name drug manufacturers and generic companies, which is especially important given that more than 80 percent of prescriptions in the United States are currently dispensed as generic drugs. “Now, with the generic industry having grown up, most people are taking generic drugs,” she said in a telephone interview. “It’s really time to level the playing field.”
The rule would also pave the way for lawsuits from patients who could now claim that generic companies did not sufficiently warn them of a drug’s dangers. In 2011, the Supreme Court ruled that such lawsuits were not valid because generic companies were required to use the same label warnings as brand-name manufacturers and thus could not be blamed for failing to warn patients about the risks of taking their drugs.
Consumer advocates, who have long called on the agency to make such a change, praised the decision because they said the current situation was unfair to patients harmed by generic drugs.
“This is an important consumer protection provision,” said Representative Chris Van Hollen, Democrat of Maryland, who had supported such a change. “It’s a long time in coming.”
The Generic Pharmaceutical Association, the industry trade group, said it was still reviewing the regulation but was concerned that the change could create confusion. In a statement, Ralph G. Neas, the association’s president, said the group was “very concerned that multiple versions of critical safety information would lead to unnecessary confusion and uncertainty for prescribers and other health care professionals, with harmful consequences for patients.”
Mr. Neas also raised the question of whether the rule was legal. “The Supreme Court has repeatedly held that generic pharmaceutical manufacturers must duplicate the language on the brand pharmaceutical manufacturer’s labels and cannot make changes to a label without F.D.A. approval,” he said in the statement. “Therefore, the agency’s authority to enact a rule that differs from the federal law is unclear.”
The proposed rule, which is open to public comment for 60 days, would set up a website that would place all updates for a given drug in one place, minimizing such confusion, Dr. Woodcock said.
The agency said the Supreme Court decision, Pliva v. Mensing, altered the incentives for generic drug companies to conduct adequate monitoring of their drugs because it eliminated the threat of lawsuits from patients who were harmed by those products. In proposing the rule, the agency said it “intended to ensure that generic drug companies actively participate with F.D.A. in ensuring the timeliness, accuracy and completeness of drug safety labeling.”
Dr. Woodcock said she did not expect that generic companies would update their labels frequently, because by the time drugs become available as generics, they have been on the market for years. “It’s not to say that we won’t learn new things, but it becomes less likely to come across new serious adverse events,” she said. Read More & Comment...To address (one hesitates to say “celebrate”) American Diabetes month, here is the latest addition to Pfizer’s Value of Medicines series -- The Value of Medicine for Type 2 Diabetes.
The development of medicines is complicated -- explaining their value should not be. Pfizer's straightforward efforts put the facts in perspective -- and in plain English. Their Value of Medicines series provides highly valuable tools for healthcare policy professionals, practitioners, and patients. Additional materials can be found here.
Knowledge is Power.
From the pages of the Oregon StatesmanJournal
From Eugene to Eugenics: Oregon's new cost-cutting strategy is to deny care to cancer patients
In an ad for the Affordable Care Act, local folk singer Laura Gibson plucks her guitar and muses about how the “Oregon way” is to “care for each one, every daughter and son.” Though a little corny, the song delivers a worthy message: Oregon is committed to providing everyone access to health care.
The commercial is part of Oregon’s new ad campaign to boost enrollment in the newly created “Cover Oregon” health insurance exchange.
But the reality strums a different, far less organic tune. Even as Oregon drops $3.2 million to spread their “each one” message, the Beaver State is also taking measures to that would deny life-saving treatments to desperately sick citizens.
In August, Oregon’s Health Evidence Review Commission issued an update to its guidelines for providing cancer treatment to low-income individuals covered by the state Medicaid program. These new guidelines require that Medicaid deny coverage for certain cancer treatments for patients that have been deemed “too” sick, haven’t responded well to previous treatments, or can’t care for themselves.
Through these new rules, Oregon state bureaucrats are severely restricting access to care and dooming potentially thousands of local patients to a premature death.
What’s worse it that these new Medicaid guidelines are not grounded in the medical literature or best clinical practices, according to Kenneth Thorpe, chairman of the Partnership to Fight Chronic Disease. Rather, according to Thorpe, they’re based “on the odds of survival observed in a group of patients.”
It’s true that for some late-stage cancer patients, the odds are long than any additional treatment can help. But without access to the latest that medical science has to offer, a patient’s survival rate simply drops to zero.
As B.J. Cavnor of the Northwest Patient Education Network powerful puts it: “Cutting patients off from cures means patients who could have beaten their illness will no longer have that chance.”
It’s a frightening move from Eugene to eugenics. These guidelines dictate that Medicaid only provide “palliative” care – painkillers, acupuncture treatments, wheelchairs, drugs for nausea, and the like.
So while Oregon won’t let Medicaid patients have access to cancer medicines that could prolong or save their lives, it will pay to make their deaths slightly less painful. Is that what Oregon considers compassionate care?
About 19,000 Oregonians are diagnosed with cancer each year. Over 640,000 state residents are covered by Medicaid -- that’s about one in five of the total state population. And the state Medicaid ranks will swell next year, when the Affordable Care Act will raise the program’s income threshold up to 138 percent of the federal poverty line.
Oregon’s new Medicaid guidelines take treatment decisions out of the hands of doctors and patients and put them in the hands of distance state bureaucrat willing to cut costs no matter the human toll. It’s the practice of cost-centric controls over patient-centric care.
Even supporters of the President’s healthcare law have taken to calling these treatment restrictions a death knell for poor cancer patients.
Cavnor, the patient advocate, has described them as “extremely frustrating and morbidly ironic, especially for those of us who have tried to argue that the Affordable Care Act doesn’t allow for ‘death panels.’”
Is this really change we can believe in? Promising to expand access to health care to all while denying it to those who need it most is brazen hypocrisy. Oregon should expect more from itself.
At the end of her song boosting the state’s health exchange, Laura Gibson sings “live long, Oregon.” That’s a good aspiration. Oregon’s state bureaucrats should live up to it.
Peter J. Pitts, a former associate commissioner at the FDA, is president of the Center for Medicine in the Public Interest.
Read More & Comment...
Substandard Subcontinent.
On its Tuesday broadcast, the CBS Evening News reported on former Ranbaxy executive Dinesh Thakur, who “was asked by his boss to investigate allegations of fraud at the company” in 2004. He soon “uncovered disturbing problems with the data required by the FDA to prove the effectiveness of Ranbaxy drugs.” According to Thakur, the company had “gotten approvals from the FDA to sell drugs that were based on no data, or data that was fraudulent.” After presenting his finding to Ranbaxy executives in 2005, Thakur “says nothing was done,” so he “blew the whistle to the FDA.”
The CBS News website reports the FDA investigation uncovered a “persistent ... pattern” by Ranbaxy of submitting “untrue statements.” Auditors found that the applications of at least 15 new generic drugs contained over 1,600 data errors, leading the FDA to conclude the company’s products were “potentially unsafe and illegal to sell.” The investigation that was launched in the wake of “Thakur’s allegations led Ranbaxy to plead guilty to seven felonies.”
Read More & Comment...I’ve just returned from Kiev, Moscow, and Algiers where I spoke on pharmaceutical quality, bioequivalence, and generic interchangability. (And, yes, I did get a chance to tour around a little too.)
Two comments worth sharing.
* In Algiers, the head of the domestic generic drugs manufacturers, told me that the issues of generic quality and therapeutic substitution were “obsolete.” It was a laugh line – because that’s how the audience responded.
* In Moscow, at a meeting of federal and provincial healthcare officials, one comment (relative to patient choice vs. limited formulary options) was, “The only patients who give us trouble are those who want more expensive treatments.”
In Russia -- if you like your insurance, you can keep it. If you don't like it, you can keep it ... quiet.
Think of it as a healthcare safety nyet.
The themes for the Algerian Society of Pharmacy’s 22nd Journées Pharmaceutiques Nationales was health policy , intellectual property , self-medication and hospital pharmacy. A panel of national and international experts addressed the generic drugs and the pharmaceutical industry in Algeria.
According to Prof. P. J. Pitts , president of the U.S. Center for Medicine in the Public Interest, "We cannot replace quality with cost. Even a high quality generic does not always give the same therapeutic effect as the innovative product . The authorities should work with physicians and pharmacists to ensure that patients remain at the center of debate. The most expensive drug is the one that does not work. The most effective treatment is one that delivers good patient outcomes," he said.
The President of the National Council of the Order of Pharmacists , Dr. Lotfi Benbahmed, insisted on continuing education for pharmacists and also the establishment of a system to determine the traceability of drugs to determine responsibility.
"The door is open to counterfeiting and quackery. It is a great danger to public health if official medication is not controlled."
The complete article (in French) can be found here.
Read More & Comment...A small item from BioCentury that deserves more attention:
Drug companies will be able to provide copay assistance to consumers who purchase health insurance through the Affordable Care Act's exchanges. HHS Secretary Kathleen Sebelius announced the decision in a letter sent on Wednesday to Rep. Jim McDermott (D-Wash.), resolving a major uncertainty about the healthcare law. Under federal kickback laws, drug companies are not permitted to provide copay assistance directly to consumers insured by federal programs such as Medicare and Medicaid. According to Sebelius' letter, HHS does not consider qualified health plans purchased through federal or state exchanges to be federal health programs.
Read More & Comment...Washington, DC—HR Policy Association, representing the chief human resource officers of more than 350 of the largest private sector employers in the United States, announces the selection of Tevi Troy, former Deputy Secretary of the U.S. Department of Health and Human Services, to lead the development of a health care initiative of the HR Policy Foundation.
HR Policy Association member companies, all of whom are large employers, collectively spend more than $75 billion annually on health care in the U.S. and are in the process of assessing their company's current and future health care strategies in light of the growing instability of the American health care system. They are seeking to establish a leadership organization dedicated to developing a more sustainable health care delivery system for the employees, dependents and retirees of large employers.
Dr. Troy is currently a Senior Fellow at Hudson Institute, where he will remain an Adjunct Fellow, and a writer and consultant on health care and domestic policy. In 2007, he was unanimously confirmed by the U.S. Senate as the Deputy Secretary of the U.S. Department of Health and Human Services. He was the chief operating officer of the largest civilian department in the federal government, with a budget of $716 billion and over 67,000 employees. In that position, he oversaw all operations, including Medicare.
HR Policy Association CEO Jeffrey C. McGuiness said, “We are delighted to announce that Dr. Troy will be leading this critically important initiative. He brings not only a wealth of understanding and expertise about health care, policy and the government, but a strong managerial and operations background. Having successfully run the largest department in the government in terms of employees and budget, focused on all aspects of health care, Dr. Troy is uniquely qualified to help guide and lead this new organization.”
Dr. Troy has extensive White House and Capitol Hill experience. During the George W. Bush Administration, he served as Deputy Assistant and then Acting Assistant to the President for Domestic Policy and was the White House’s lead adviser on health care, labor, education, transportation, immigration, crime, veterans and welfare. Dr. Troy also served as the Policy Director for U.S. Senator John Ashcroft and as Senior Domestic Policy Adviser and Domestic Policy Director for the U.S. House Policy Committee.
In addition to his senior level government work and health care expertise, Dr. Troy is also an author and presidential historian. He is a frequent television and radio analyst, and has appeared on CNN, CNBC, FOX News, FOX Business, and The Jim Lehrer Show, among other outlets. He is currently on tour for his latest book, What Jefferson Read, Ike Watched, and Obama Tweeted: 200 Years of Popular Culture in the White House and is the author of Intellectuals and the American Presidency: Philosophers, Jesters, or Technicians? (Lanham: Rowman & Littlefield, 2002). He has written over 100 articles, for The Wall Street Journal, The Washington Post, Forbes, The New Republic, Commentary, Reason, Investor’s Business Daily, National Review, Washingtonian, The Weekly Standard, and other publications. Dr. Troy has a B.S. in Industrial and Labor Relations from Cornell University and an M.A and Ph.D. in American Civilization from the University of Texas at Austin.
Read More & Comment...I'm in town to give a presentation on the importance of API and excipient quality, the urgency of advancing pharmacovigilance, and the evolution of both regulatory innovation and international regulatory fraternity.
The local english-language newspaper is The Moscow Times. It's what you get at the hotel. And amidst all the stories on NSA wire-tapping was an editorial that nearly made my heart stop. The title, "Dispelling the Smoke Screen." Here's the opening paragraph:
"The statement that smoking is harmful to your health is only a theory, not a medical fact."
Don't believe me? Read the article and then exhale. And then get angry.
This in a nation where non-communicable diseases (most notably alchohol and heart disease) are major killers. 50% of Russians smoke cigarettes.
I intend to discuss this article during my presentation.
Outrageous! Read More & Comment...
Check out Pfizer’s new “Value of Medicines: How Medicines Have Changed Our World” position papers. They’re actually concise and to the point (unlike many of the policy papers we see these days).
At the moment there are three to choose from:
* The Value of Statins
* The Value of Oncology Medicines
* The Value of Adherence
They can all be found here.
According to the website, “Papers will soon be available demonstrating value in a variety of areas including Vaccines, Diabetes, Rare Diseases, Atrial Fibrillation-Stroke, Pain, Renal Cell Carcinoma, Smoking Cessation, Breast Cancer, and more. Stay tuned!”
Kudos to Ian & Company for a job well done.
Read More & Comment...According to Friedrich Hayek, socialists are wrong because they disregard the fact that modern civilization naturally evolved and was not planned. Hayek refers to this fundamental error as “the fatal conceit.”
And on CNBC it’s what Larry Kudlow used to describe ObamaCare during a debate on the roll-out of the Affordable Care Act– the fatal conceit. The panel consisted of Jonathan Gruber (MIT professor and a designer of the ACA), Matt Welch (editor of Reason Magazine), and me.
Here are two clips to enjoy:
“ObamaCare already a huge turkey”
“Previous system of healthcare archaic”
Are we on a slippery slope to a single payer system? If we are, you can kiss investment goodbye, along with innovation, and any hopes for the future of personalized medicine. Is this a system that was designed to fail?
You be the judge.
We shall not grow wiser before we learn that much we have done was very foolish
-- Friedrich Hayek
Read More & Comment...Factory Closing Means One Less Producer
Nick Mulcahy, MedScape
The already small number of producers of generic cancer drugs in the United States just got smaller.
Ben Venue Laboratories, a major manufacturer of generic chemotherapy injectables, announced the closure of its plant, in Bedford, Ohio, earlier this month, citing problems with both the facility and projected revenues. It will effectively depart the US market by the end of the year.
The loss is a significant blow to the recovery of the US marketplace for generic cancer drugs, said Erin Fox, PharmD, director of the drug information service at the University of Utah Hospitals and Clinics in Salt Lake City. Dr. Fox has been monitoring drug shortages in the United States since 2001.
"I really feel like it's a big step back," she told Medscape Medical News in an interview.
Our supply chain is that much more fragile.
The US market for generic cancer chemotherapy injectables has improved since the "crisis" of 2011, she said. But the loss of Ben Venue, which a company Web site touts as "one of the largest sterile injectable facilities in the world," is important. "Our supply chain is that much more fragile with the closing," said Dr. Fox.
"A significant portion of the ability of the market to meet demand is now gone," said William Greene, PharmD, chief pharmaceutical officer at St. Jude Children's Research Hospital in Memphis, Tennessee, who also spoke to Medscape Medical News about the Ben Venue closure.
"We are reaching a scary situation," said Sara Butler, PharmD, oncology clinical pharmacy supervisor at Barnes-Jewish Hospital in St. Louis, Missouri, about the loss of manufacturing capacity in an interview.
Another expert said that the impact of the plant closure is unclear.
"There's not enough transparency [from product distributors about the factory origins of generic drugs] to know exactly what [Ben Venue is] producing at this point," said Jeffrey Ward, MD, from the Swedish Medical Center in Seattle, who is chair of the Clinical Practice Committee of the American Society of Clinical Oncology (ASCO). "That's almost scarier than knowing what the impact is," he told Medscape Medical News.
Set Back of 4 to 6 Years
Before the news of the Ben Venue closing, Dr. Fox believed that the problem would see a significant improvement in ongoing and active shortages in about 2 years. Now, that time period doubles or triples. "I think it does set us back 4 to 6 years," she said.
Dr. Fox's speculative timeline was revealed earlier this week in the trade publication The Cancer Letter.
Ben Venue, which specializes in making injectable drugs, has been a major player in the US market for generic chemotherapy, Dr. Fox explained. There is no new manufacturer to take Ben Venue's place. "Nobody's jumping on board right now," she said.
We really need another supplier.
In the US cancer generics market, there are 3 "workhorse companies" that provide most of the generic chemotherapy injectables, said Dr. Fox. These are Hospira, Teva, and Bedford Laboratories, which is a distributor of Ben Venue products and products of other third parties. (Bedford and Ben Venue are both owned by Boehringer Ingelheim.) Pfizer is also in the market. Given this small circle, the loss of Ben Venue's manufacturing is significant, she believes. "We really need another supplier [of generic oncology products]," she said.
However, she emphasized that the situation is not as bad as it was 2 years ago. "People are not going without treatment, as in 2011."
A different opinion comes from Dr. Butler, who said that at Barnes-Jewish Hospital, there are still times when staff have to tell cancer patients that "we don't have certain drugs." She believes the situation has become "much worse in the last 2 years."
New Data on Chemotherapy Shortages
Currently, there are 31 cancer drugs actively in short supply, which is down from nearly 40 in 2011, according to Dr. Fox.
Also, there were only 4 chemotherapies that newly went into shortage in 2013, compared with 26 new chemotherapy shortages in 2011, according to data from a presentation that Dr. Fox made this week at a conference of hospital executives in Atlanta.
However, chemotherapies in short supply are just a small part of the overall drug shortage picture in the United States, Dr. Fox pointed out.
Notably, Ben Venue played a role in the cancer drug shortage crisis of 2011, Dr. Fox believes.
The production facility closed that year after a series of customer complaints about products and inspections from the US Food and Drug Administration (FDA) and other agencies. Subsequently, 2 very important mainstays of cancer treatment — doxorubicin and methotrexate — went into extremely short supply, said Dr. Fox. It turned out that Ben Venue was a producer of both drugs.
The marketplace eventually reacted, with some help from the FDA, and both dire shortages were alleviated.
Among other events, the FDA approved APP Pharmaceuticals as the manufacturer of a preservative-free form of methotrexate, and allowed an Indian manufacturer, Sun Pharma Global, to export generic doxorubicin to the United States.
After the 2011 closing, the Ben Venue plant resumed "limited" production in 2012. By that time, the drastic impact of the initial closure on the marketplace had passed, said Dr. Fox. "All of that pain has been dealt with in one way or another," she explained, adding that it holds true today, even with the news of the plant closure.
Immediate Problems
The closure nonetheless creates immediate clinical problems. For instance, Ben Venue and its sibling company, Bedford Laboratories, have been the sole US suppliers of thiotepa, Dr. Fox pointed out. The FDA is now allowing thiotepa, which is used in stem cell transplants and other settings, to be imported from Italy. "But that's very inconvenient for people; there's almost a month's delay," she said.
Dr. Greene said that procuring thiotepa from abroad is "exceedingly expensive and time consuming," and that St. Jude's is now looking for alternative drugs for chemotherapy regimens involving thiotepa. He is also concerned that daunorubicin, which is used in the treatment of leukemias, will fall into short supply because, now, only Teva will be producing the drug.
The Ben Venue plant has also been the world's sole manufacturer of Doxil, the branded version of doxorubicin hydrochloride liposome injection, which is owned by Johnson & Johnson. The product could disappear from the market for a time until a new manufacturer is found, as reported this week by Medscape Medical News.
Currently, Doxil is being evaluated in a number of clinical trials. Its potential disappearance from the marketplace puts the viability of these trials at risk because a generic substitution, which would be acceptable for patients in the clinic, is not possible in a research setting, ASCO's Dr. Ward pointed out.
The Murky World of Generics Manufacturing
Dr. Ward said he was surprised to learn that a branded drug, Doxil, owned by a major drug company, Janssen/Johnson & Johnson, was made by a generics manufacturer. But the cancer generics market is full of surprises and unknowns, he noted.
Take the example of Bedford Laboratories, which began as a division of Ben Venue in 1993. Both entities have been properties of Boehringer Ingelheim since 1997.
Unlike Ben Venue, Bedford will remain in business.
That's good news, because Bedford is the leading provider of cancer generic sterile injectables in the United States, with 30% of the market, according to an analysis led by Janet Woodcock, MD, head of the pharmaceuticals division at the FDA, and published earlier this year (Clin Pharmacol Ther. 2013;93:170-176).
But currently, Bedford is "out of stock" of a long list of sterile injectables, including many oncology products. The list, posted October 1 on the company Web site, includes Adriamycin, cytarabine, dacarbazine, etoposide, gemcitabine, methotrexate, paclitaxel, and vinblastine.
Some of these are very important products, said Dr. Ward. For instance, gemcitabine is part of the standard of care in pancreatic cancer.
"What you don't know from that list is how many of these drugs were manufactured by Ben Venue and how many come from elsewhere," he explained.
The company product announcement says these out-of-stock injectable drugs could be available "pending production as capacity permits."
"Bedford will continue to distribute products manufactured by Ben Venue Laboratories until that inventory is depleted," a company spokesperson said in an email to Medscape Medical News.
Generally, "there is not much transparency in this market," observed Dr. Ward.
He explained that producers of generic sterile injectables in the United States are regulated in terms of original licensure and the design of manufacturing processes. "But the FDA does not know who is having problems with a factory or profitability," he said, referring to the 2 reasons cited by Ben Venue for leaving the US market.
Generics companies can also switch their manufacturing from a drug that is not profitable to one that is, which can play havoc with the market availability of agents, said Dr. Ward.
He noted that the generics market, which includes chemotherapy injectables, is inherently volatile because profit margins are thin. Once on the market, "generics get very cheap very quickly," he said. Any variable that increases costs cuts into these already thin profits. This is exacerbated by that fact that the Centers for Medicare & Medicaid Services only updates the stated average sales price every 6 months, forcing companies to wait long periods for an approved adjustment in price. As a result, companies can feel a need to start and stop the production of certain products.
St Jude's Dr. Greene agrees that the generics market is a murky business in which many manufacturing and related planning details are not disclosed. "We just know when something is gone," he noted. The FDA Safety and Innovation Act, enacted in 2012, requires that drug manufacturers to notify the FDA as soon as they anticipate interruptions in drug production, and 6 months in advance if a product is to be discontinued. Although the law "improves the situation," Dr. Greene said, it has shortcomings.
Dr. Ward observed that the generics manufacturers managed to retain a number of loopholes in the law, which weaken its sentinel nature.
Seven companies supply 90% of the generic injectables market, said Dr. Fox. However, not all 7 make oncology products. Who manufacturers what product at what time is not publicly available information, and must be discerned through data detective work, she said.
The whole model is problematic, said Dr. Ward. "We have left it to business to ensure our drug supply."
Read More & Comment...
Medical errors are a real problem. I won’t deny that.
It was bad enough when the often-quoted Institute of Medicine figure that 98,000 deaths per year in the US are caused by medical errors was in vogue, but now a paper in the Journal of Patient Safety states that adverse medical events result in 210,000 to 400,000 deaths per year and 10 to 20 times those numbers of serious harms.
Since the paper disparages the medical profession, it has received a lot of media attention.
Most articles about it simply regurgitate the dismal estimates without any real attempt to dig into the paper’s methods.
Let’s take a closer look.
As is true of many papers, the abstract is a bit sketchy when describing how the paper arrived at its conclusion.
The full text of the paper reveals the author found four studies that looked at what are described as preventable adverse events in US hospitals within the last seven years. All four used the Global Trigger Tool which involves the screening of records for adverse events by nurses or pharmacists and a secondary review by physicians.
Based on opinions by “experts,” the author made a key, but erroneous, assumption that all adverse events are preventable.
Read the full blog here.
Read More & Comment...
Social Networks
Please Follow the Drugwonks Blog on Facebook, Twitter, LinkedIn, YouTube & RSS
Add This Blog to my Technorati Favorites